VEP是国际三大数据库之一的ENSEMBL提供的,也是非常主流和方便,但它是基于perl语言的,所以在模块方面可能会有点烦人。跟snpEFF一样,也是对遗传变异信息提供更具体的注释,而不仅仅是基于位点区域和基因。如果你熟悉外显子联盟这个数据库EXAC(ExAC.r0.3.sites.vep.vcf.gz),你可以下载它所有的突变记录数据,看看它对每个变异位点到底注释了些什么,它就是典型的用VEP来注释的。 Continue reading
五
05
VEP是国际三大数据库之一的ENSEMBL提供的,也是非常主流和方便,但它是基于perl语言的,所以在模块方面可能会有点烦人。跟snpEFF一样,也是对遗传变异信息提供更具体的注释,而不仅仅是基于位点区域和基因。如果你熟悉外显子联盟这个数据库EXAC(ExAC.r0.3.sites.vep.vcf.gz),你可以下载它所有的突变记录数据,看看它对每个变异位点到底注释了些什么,它就是典型的用VEP来注释的。 Continue reading
这个软件比较重要,尤其是对做遗传变异相关研究的,很多人做完了snp-calling后喜欢用ANNOVAR来进行注释,但是那个注释还是相对比较简单,只能得到该突变位点在基因的哪个区域,那个基因这样的信息,如果想了解更具体一点,就需要更加功能化的软件了,snpEFF就是其中的佼佼者,而且是java平台软件,非常容易使用!而且它的手册写的非常详细:http://snpeff.sourceforge.net/SnpEff_manual.html Continue reading
string数据库是PPI领域里面最完备已经最受欢迎的数据库了。如果直接在谷歌里面搜索PPI,映入眼帘就是string的官网,它们的主页现在是html5啦,比较精美: http://string-db.org/
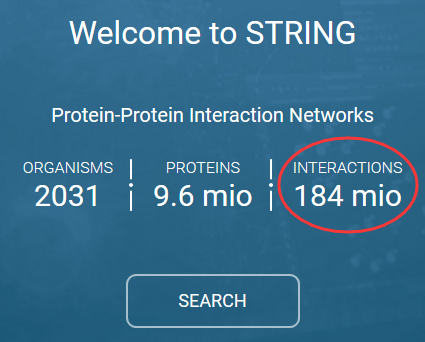
写的很霸气,近两亿的记录,不过一般大家只会关心一个物种,比如人,其实还不到一千万!
我们直接进入下载界面,找到人类的数据,人类的物种ID是9606.
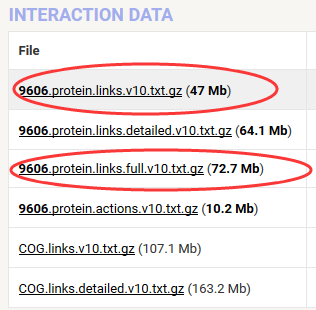
需要一定许可才能下载完整版本,我这里测试最上面那个公开版本数据!
数据很简单,就是protein+protein+score,共八百多万行记录,记录着string数据库搜集的所有可能以及可信的蛋白相互作用!但是它的蛋白ID是ENSEMBL的ID,所以需要转换成基因的ID,才能被大多数人使用,因为大家的研究单位一般是基因,所以蛋白相互作用略等于基因相互作用。
基因ID转换,我推荐用org.Hs.eg.db这个R的包,很容易就可以实现的!
> tmp=toTable(org.Hs.egENSEMBLPROT) > dim(tmp) [1] 110916 2 > head(tmp) gene_id prot_id 1 1 ENSP00000263100 2 1 ENSP00000470909 3 2 ENSP00000443302 4 2 ENSP00000323929 5 2 ENSP00000438599 6 2 ENSP00000445717 |
|
|
有约500多个蛋白ID是无法转换成对应的基因的,这个很正常,毕竟这种ID本来就不稳定,很多用着用着就失效了!
转换好之后就可以上传到数据库啦,然后可以供其它可视化或者分析程序使用!
前面讲到affy处理的芯片平台是有限的,一般是hgu 95系列和133系列,[HuGene-1_1-st] Affymetrix Human Gene 1.1 ST Array这个平台虽然也是affymetrix公司的,但是affy包就无法处理 了,这时候就需要oligo包了!
oligo包是R语言的bioconductor系列包的一个,就一个功能,读取affymetix的基因表达芯片数据-CEL格式数据,处理成表达矩阵!!!
Affymetrix的探针(proble)一般是长为25碱基的寡聚核苷酸;探针总是以perfect match 和mismatch成对出现,其信号值称为PM和MM,成对的perfect match 和mismatch有一个共同的affyID。
CEL文件:信号值和定位信息。
CDF文件:探针对在芯片上的定位信息
affy包是R语言的bioconductor系列包的一个,就一个功能,读取affymetix的基因表达芯片数据-CEL格式数据,处理成表达矩阵!!!
回归的本质是建立一个模型用来预测,而逻辑回归的独特性在于,预测的结果是只能有两种,true or false
在R里面做逻辑回归也很简单,只需要构造好数据集,然后用glm函数(广义线性模型(generalized linear model))建模即可,预测用predict函数。
我这里简单讲一个例子,来自于加州大学洛杉矶分校的课程
以前我写过如何使用GEOquery和GEOmetadb, 它们的确很强大,也很好用,做芯片数据pipeline的时候可以省很多力,但最近很多朋友都反应它联网有问题,经常无法下载数据!
为了解决这个问题,我仔细又研究了一下GEO数据库,其实官网本身就提供了WEB API接口,直接根据需求定制化下载数据!
我们使用GEO数据,无非就是想根据study ID号(比如:GSE1009)得到它的raw CEL文件,或者表达矩阵,或者样本分组信息!!!
如果用R包GEOquery来完成这个目的,请参考我的说明书:
其实raw CEL文件,直接自己拼接url即可
ftp://ftp.ncbi.nlm.nih.gov/geo/series/GSE1nnn/GSE1009/matrix/GSE1009_series_matrix.txt.gz
##表达矩阵,需要用在R里面read,skip掉注释信息,tab键分割
ftp://ftp.ncbi.nlm.nih.gov/geo/series/GSE1nnn/GSE1009/suppl/GSE1009_RAW.tar
##芯片原始数据,用affy包来读取
http://www.ncbi.nlm.nih.gov/geo/browse/?view=samples&series=1009&mode=csv
###样本分组信息
根据任意study ID号,非常容易就可以拼接出这些url,完全hold住GEOquery这个包的所有功能!
如果该研究涉及到的样本较多,你还可以根据下面的文件列表来有选择性的抓取样本!
ftp://ftp.ncbi.nlm.nih.gov/geo/series/GSE1nnn/GSE1009/suppl/filelist.txt
你要明白的就是浏览器的get请求而已,把下面的字符串组合成一个完整的URL即可
view=series& ## 四种,zsort=date&mode=csv& ##很重要,可以直接下载csv文件page=$i&display=5000 ##很重要查看总数:curl --silent "http://www.ncbi.nlm.nih.gov/geo/browse/" | grep "total_count"
limma真不愧是最流行的差异分析包,十多年过去了,一直是芯片数据处理的好帮手。
现在又可以支持RNA-seq数据,我赶紧试用了一下!
我下面只讲用法,大家看代码就明白了!
最近经常出现一个错误,类似于package ‘airway’ is not available (for R version 3.1.0)
就是某些包在R的仓库里面找不到,这个错误非常普遍,stackoverflow上面非常详细的解答:
在阅读这个答案的时候,我发现了一个非常有用的函数!available.packages()可以查看自己的机器可以安装哪些包!
我以前写过DESeq,以及过时了:http://www.bio-info-trainee.com/867.html
正好准备筹集bioconductor中文社区,我写简单讲一下DESeq2这个包如何用!
感谢读者的指正,我以前写的一个程序是错的,从算法设计上就错了!
http://www.bio-info-trainee.com/926.html
我从新设计了一个算法,经过再三检查,我可以确信它是对的,至于是否高效,就不敢保证了,也希望有更多热心的读者帮助我改正,或者跟我讨论,请直接联系我的邮箱jmzeng1314 at(防爬虫) 163.com
dbSNP的ID直接在NCBI的dbSNP官网可以看到详细介绍,现在已经更新到146版本了,一般人看到一个ID肯定什么信息都获取不到,毕竟这只是人家NCBI规定的一个ID而已。但是HGVS突变形式就有非常详细的信息了。
人类基因组变异协会(HGVS)官方组织规定了mutation该如何记录:http://www.hgvs.org/mutnomen/recs.html 推荐大家都仔细阅读!!!
大家分析生物信息学数据的时候必不可少的步骤就是利用各种公共资源对自己的数据进行注释。
这时候可能会用到mysql,把一些公共数据库本地化,方便使用,但是数据的下载已经存储到mysql等数据库中间会有很多值得玩味的事情。
我这里给大家指出一个还算比较标准的参考,就是bioconductor官方制作的数据库设计代码。
bioconductor官方注释方面的包(主要是各种ID的转换,KEGG或者GO这样的功能注释,基因信息注释,转录本,外显子起始终止等等)
目前为止,bioconductor是3.3版本,共896个包
大部分包都是以sqlite的数据库标准发布,所以建表语句是一样的。
所有代码见:https://github.com/Bioconductor-mirror/AnnotationDbi/blob/release-3.2/inst/DBschemas
部分代码如下:
CREATE TABLE metadata (
name VARCHAR(80) PRIMARY KEY,
value VARCHAR(255)
);
CREATE TABLE go_ontology (
ontology VARCHAR(9) PRIMARY KEY, -- GO ontology (short label)
term_type VARCHAR(18) NOT NULL UNIQUE -- GO ontology (full label)
);
CREATE TABLE go_term (
_id INTEGER PRIMARY KEY,
go_id CHAR(10) NOT NULL UNIQUE, -- GO ID
term VARCHAR(255) NOT NULL, -- textual label for the GO term
ontology VARCHAR(9) NOT NULL, -- REFERENCES go_ontology
definition TEXT NULL, -- textual definition for the GO term
FOREIGN KEY (ontology) REFERENCES go_ontology (ontology)
);
CREATE TABLE sqlite_stat1(tbl,idx,stat);
CREATE TABLE go_obsolete (
go_id CHAR(10) PRIMARY KEY, -- GO ID
term VARCHAR(255) NOT NULL, -- textual label for the GO term
ontology VARCHAR(9) NOT NULL, -- REFERENCES go_ontology
definition TEXT NULL, -- textual definition for the GO term
FOREIGN KEY (ontology) REFERENCES go_ontology (ontology)
);
最流行的差异分析软件就是limma了,它现在更新了一个voom的算法,所以既可以对芯片数据,也可以对转录组高通量测序数据进行分析,其它所有的差异分析软件其实都是模仿这个的。
我以前讲到过做差异分析,需要三个数据:
前面两个肯定是必须的,有表达矩阵,样本必须进行分组,才能分析,但是我看到过好几种例子,有的有差异比较矩阵,有的没有。
后来我仔细研究了一下limma包的说明书,发现这其实是一个很简单的问题。
library(CLL)
data(sCLLex)
library(limma)
design=model.matrix(~factor(sCLLex$Disease))
fit=lmFit(sCLLex,design)
fit=eBayes(fit)
options(digits = 4)
#topTable(fit,coef=2,adjust='BH')
> topTable(fit,coef=2,adjust='BH')
logFC AveExpr t P.Value adj.P.Val B
39400_at 1.0285 5.621 5.836 8.341e-06 0.03344 3.234
36131_at -0.9888 9.954 -5.772 9.668e-06 0.03344 3.117
33791_at -1.8302 6.951 -5.736 1.049e-05 0.03344 3.052
1303_at 1.3836 4.463 5.732 1.060e-05 0.03344 3.044
36122_at -0.7801 7.260 -5.141 4.206e-05 0.10619 1.935
36939_at -2.5472 6.915 -5.038 5.362e-05 0.11283 1.737
41398_at 0.5187 7.602 4.879 7.824e-05 0.11520 1.428
32599_at 0.8544 5.746 4.859 8.207e-05 0.11520 1.389
36129_at 0.9161 8.209 4.859 8.212e-05 0.11520 1.389
37636_at -1.6868 5.697 -4.804 9.355e-05 0.11811 1.282
library(CLL)
data(sCLLex)
library(limma)
design=model.matrix(~0+factor(sCLLex$Disease))
colnames(design)=c('progres','stable')
fit=lmFit(sCLLex,design)
cont.matrix=makeContrasts('progres-stable',levels = design)
fit2=contrasts.fit(fit,cont.matrix)
fit2=eBayes(fit2)
options(digits = 4)
topTable(fit2,adjust='BH')
logFC AveExpr t P.Value adj.P.Val B
39400_at -1.0285 5.621 -5.836 8.341e-06 0.03344 3.234
36131_at 0.9888 9.954 5.772 9.668e-06 0.03344 3.117
33791_at 1.8302 6.951 5.736 1.049e-05 0.03344 3.052
1303_at -1.3836 4.463 -5.732 1.060e-05 0.03344 3.044
36122_at 0.7801 7.260 5.141 4.206e-05 0.10619 1.935
36939_at 2.5472 6.915 5.038 5.362e-05 0.11283 1.737
41398_at -0.5187 7.602 -4.879 7.824e-05 0.11520 1.428
32599_at -0.8544 5.746 -4.859 8.207e-05 0.11520 1.389
36129_at -0.9161 8.209 -4.859 8.212e-05 0.11520 1.389
37636_at 1.6868 5.697 4.804 9.355e-05 0.11811 1.282
大家运行一下这些代码就知道,两者结果是一模一样的。
而差异比较矩阵的需要与否,主要看分组矩阵如何制作的!
design=model.matrix(~factor(sCLLex$Disease))
design=model.matrix(~0+factor(sCLLex$Disease))
有本质的区别!!!
前面那种方法已经把需要比较的组做出到了一列,需要比较多次,就有多少列,第一列是截距不需要考虑,第二列开始往后用coef这个参数可以把差异分析结果一个个提取出来。
而后面那种方法,仅仅是分组而已,组之间需要如何比较,需要自己再制作差异比较矩阵,通过makeContrasts函数来控制如何比较!
这是我们bioconductor中文社区的一个简单测试好像放在博客里面markdown的语法除了问题,欢迎直接去github查看
这个对象其实是对表达矩阵加上样本分组信息的一个封装,由biobase这个包引入。它是eSet这个对象的继承。
下面是一个具体的例子,来源于CLL这个包,是用hgu95av2芯片测了22个样本
> library(CLL)
> data(sCLLex)
> sCLLex
ExpressionSet (storageMode: lockedEnvironment)
assayData: 12625 features, 22 samples ##表达矩阵
element names: exprs
protocolData: none
phenoData
sampleNames: CLL11.CEL CLL12.CEL ... CLL9.CEL (22 total)
varLabels: SampleID Disease ## 样本分组信息
varMetadata: labelDescription
featureData: none
experimentData: use 'experimentData(object)'
Annotation: hgu95av2
> exprMatrix=exprs(sCLLex)
> dim(exprMatrix)
[1] 12625 22
> meta=pData(sCLLex)
> table(meta$Disease)
progres. stable
14 8
>
根据上面的信息可以看出该芯片共12625个探针,这22个样本根据疾病状态分成两组,14vs8
这个数据对象就可以打包做很多包的分析输入数据。
对这个包的分析,重点就是 `exprs` 函数提取表达矩阵,`pData` 函数看看该对象的样本分组信息。
下面这个例子充分说明了
ExpressionSet对象的重要性
> library(limma)
> design=model.matrix(~factor(sCLLex$Disease))
> fit=lmFit(sCLLex,design)
> fit=eBayes(fit)
> options(digits = 4)
> topTable(fit,coef=2,adjust='BH')
logFC AveExpr t P.Value adj.P.Val B
39400_at 1.0285 5.621 5.836 8.341e-06 0.03344 3.234
36131_at -0.9888 9.954 -5.772 9.668e-06 0.03344 3.117
33791_at -1.8302 6.951 -5.736 1.049e-05 0.03344 3.052
1303_at 1.3836 4.463 5.732 1.060e-05 0.03344 3.044
36122_at -0.7801 7.260 -5.141 4.206e-05 0.10619 1.935
36939_at -2.5472 6.915 -5.038 5.362e-05 0.11283 1.737
41398_at 0.5187 7.602 4.879 7.824e-05 0.11520 1.428
32599_at 0.8544 5.746 4.859 8.207e-05 0.11520 1.389
36129_at 0.9161 8.209 4.859 8.212e-05 0.11520 1.389
37636_at -1.6868 5.697 -4.804 9.355e-05 0.11811 1.282
>
还有非常多的其它包会使用 ExpressionSet 对象,我就不一一介绍了。
ExpressionSet 对象根据上面的讲解,我们知道了在这个对象其实很简单,就是对表达矩阵加上样本分组信息的一个封装。 所以我们就用上面得到的exprMatrix和meta来构建一个ExpressionSet对象,biobase包里面提供了详细的说明,建议大家仔细看官方手册
metadata <- data.frame(labelDescription=c('SampleID', 'Disease'),
row.names=c('SampleID', 'Disease'))
phenoData <- new("AnnotatedDataFrame",data=meta,varMetadata=metadata)
myExpressionSet <- ExpressionSet(assayData=exprMatrix,
phenoData=phenoData,
annotation="hgu95av2")
> myExpressionSet
ExpressionSet (storageMode: lockedEnvironment)
assayData: 12625 features, 22 samples
element names: exprs
protocolData: none
phenoData
sampleNames: CLL11.CEL CLL12.CEL ... CLL9.CEL (22 total)
varLabels: SampleID Disease
varMetadata: labelDescription
featureData: none
experimentData: use 'experimentData(object)'
Annotation: hgu95av2
>
从上面的构造过程可以看出,重点就是表达矩阵加上样本分组信息
ExpressionSet 对象 library(ALL)
data(ALL)
ALL
ExpressionSet (storageMode: lockedEnvironment)
assayData: 12625 features, 128 samples
element names: exprs
protocolData: none
phenoData
sampleNames: 01005 01010 … LAL4 (128 total)
varLabels: cod diagnosis … date last seen (21 total)
varMetadata: labelDescription
featureData: none
experimentData: use ‘experimentData(object)’
pubMedIds: 14684422 16243790
Annotation: hgu95av2
这个数据非常出名,很多其它算法包都会拿这个数据来举例子,只有真正理解了ExpressionSet对象才能学会bioconductor系列包
ExpressionSet 对象 gse1009=GEOquery::getGEO("GSE1009")
gse1009[[1]] ## 这就是ExpressionSet对象
我发现糗世界讲的要比我好:http://blog.qiubio.com:8080/archives/2957
在Biobase基础包中,ExpressionSet是非常重要的类,因为Bioconductor设计之初是为了对基因芯片数据进行分析,而ExpressionSet正是Bioconductor为基因表达数据格式所定制的标准。它是所有涉及基因表达量相关数据在Bioconductor中进行操作的基础数据类型,比如affyPLM, affy, oligo, limma, arrayMagic等等。所以当我们学习Bioconductor时,第一个任务就是了解并掌握ExpressionSet的一切。
ExpressionSet的组成:
有了这些,所有实验相关的信息基本全备。
ExpressionSet继承了eSet类,属性基本和eSet保持一致。
那么,对于一个ExpressionSet,哪些属性是必须的?哪些有可能缺失呢?很显然,assayData是必须的,其它的可能会缺失,但是不能都缺失,因为那样的话就无法完成数据分析的工作。
对于ExpressionSet最重要的操作就是如何取出子集了。有时候在进行质量分析之后,我们对其中一些样品的数据不满意,想从已经实例化的ExpressionSet中抽取掉,或者我们希望对样品进行分组,都需要使用到Subset的概念。那么如何抽取子集呢?
我们可以象操作矩阵那样对其进行子集操作:vv <- exampleSet[1:5, 1:3]
使用它的一些属性来对其进行子集操作:males <- exampleSet[, exampleSet$gender == "Male"];
在R里面实现这个功能其实非常简单,难的是很多packages经常会出现安装问题,更有的人压根不看芯片平台是什么,芯片对应的package是什么,就开始到处发问,自学能力实在是堪忧!
我前面有写目前所有bioconductor支持的芯片平台对应关系:通过bioconductor包来获取所有的芯片探针与gene的对应关系
但那其实是一个很笨的办法,得到所有的各式各样的探针ID与基因的对应关系,以为它绕路了,正常情况只需要在GEO里面找到芯片对应基因关系即可,没必要下载那么多package的,但是这样做的好处也是很明显的, 对很多初学者来说,如果package能解决的话,就省心很多,比如下面这个转换关系:
suppressPackageStartupMessages(library(CLL))## 这个package自带了一个数据,是我们需要用的data(sCLLex) ## 这个数据里面有24个样本,分成两组,可以直接拿来测试差异基因分析library(hgu95av2.db) ## 一定要搞清楚自己的芯片是什么数据包exprSet=exprs(sCLLex) ##得到表达数据矩阵,但是矩阵的行名,是探针ID,无法理解,需要转换##首先你取出所有的探针ID,#这里可以用三种方法来得到symbol,或者得到entrezID也可以probeset=rownames(exprSet)Symbol=as.character(as.list(hgu95av2SYMBOL[probeset]))#annotate包提供 getSYMBOL( probeset ,"hgu95av2" )#还可以用lookUp函数 lookUp( probeset , "hgu95av2", "SYMBOL")#这些只是技巧而已啦a=cbind.data.frame(Symbol,exprSet)## 下面这个函数是对每个基因挑选最大表达量探针rmDupID <-function(a=matrix(c(1,1:5,2,2:6,2,3:7),ncol=6)){exprSet=a[,-1]rowMeans=apply(exprSet,1,function(x) mean(as.numeric(x),na.rm=T))a=a[order(rowMeans,decreasing=T),]exprSet=a[!duplicated(a[,1]),]#exprSet=apply(exprSet,2,as.numeric)exprSet=exprSet[!is.na(exprSet[,1]),]rownames(exprSet)=exprSet[,1]exprSet=exprSet[,-1]return(exprSet)}exprSet=rmDupID(a)
我是受到了SOAPfuse的启发才想到整理各种基因组版本的对应关系,完整版!!!
以后再也不用担心各种基因组版本混乱了,我还特意把所有的下载链接都找到了,可以下载任意版本基因组的基因fasta文件,gtf注释文件等等!!!
GRCh36 (hg18): ENSEMBL release_52.GRCh37 (hg19): ENSEMBL release_59/61/64/68/69/75.GRCh38 (hg38): ENSEMBL release_76/77/78/80/81/82.
Feb 13 2014 00:00 Directory April_14_2003 Apr 06 2006 00:00 Directory BUILD.33 Apr 06 2006 00:00 Directory BUILD.34.1 Apr 06 2006 00:00 Directory BUILD.34.2 Apr 06 2006 00:00 Directory BUILD.34.3 Apr 06 2006 00:00 Directory BUILD.35.1 Aug 03 2009 00:00 Directory BUILD.36.1 Aug 03 2009 00:00 Directory BUILD.36.2 Sep 04 2012 00:00 Directory BUILD.36.3 Jun 30 2011 00:00 Directory BUILD.37.1 Sep 07 2011 00:00 Directory BUILD.37.2 Dec 12 2012 00:00 Directory BUILD.37.3
1. Navigate to http://genome.ucsc.edu/cgi-bin/hgTables2. Select the following options:
clade: Mammal
genome: Human
assembly: Feb. 2009 (GRCh37/hg19)
group: Genes and Gene Predictions
track: UCSC Genes
table: knownGene
region: Select "genome" for the entire genome.
output format: GTF - gene transfer format
output file: enter a file name to save your results to a file, or leave blank to display results in the browser3. Click 'get output'.
for i in $(seq 1 22) X Y M;
do echo $i;
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/chromosomes/chr${i}.fa.gz;## 这里也可以用NCBI的:ftp://ftp.ncbi.nih.gov/genomes/M_musculus/ARCHIVE/MGSCv3_Release3/Assembled_Chromosomes/chr前缀
done
gunzip *.gz
for i in $(seq 1 22) X Y M;
do cat chr${i}.fa >> hg19.fasta;
done
rm -fr chr*.fasta
开发单位:华大,SOAP系列软件套装!
二,输入数据准备
6.5K Jun 15 2009 cytoBand.txt.gz3.0G Oct 12 2012 hg19.fa2.5M Mar 15 10:30 HGNC_Gene_Family_dataset38M Feb 8 2014 Homo_sapiens.GRCh37.75.gtf.gz202 Jan 19 16:07 HumanRef_refseg_symbols_relationship.list
文件下载地址,作者已经给出了!
这一步耗时很长,4~6小时,创造了transcript.fa和gene.fa,然后还对他们建立bwa和soap的index,所以有点慢!
Congratulations!You have constructed SOAPfuse database files successfully.These database files are all stored in directory you supplied:/home/jmzeng/biosoft/SOAPfuse/db_v1.27/They are all generated based on public data files you supplied:whole_genome_fasta_file: /home/jmzeng/biosoft/SOAPfuse/db_v1.27/raw/hg19.fagtf_annotation_file: /home/jmzeng/biosoft/SOAPfuse/db_v1.27/raw/Homo_sapiens.GRCh37.75.gtf.gzChr_Bandregion_file: /home/jmzeng/biosoft/SOAPfuse/db_v1.27/raw/cytoBand.txt.gzHGNC_gene_family_file: /home/jmzeng/biosoft/SOAPfuse/db_v1.27/raw/HGNC_Gene_Family_datasetgtf_segname2refseg_list: /home/jmzeng/biosoft/SOAPfuse/db_v1.27/raw/HumanRef_refseg_symbols_relationship.list
DB_db_dir = /DATABASE_DIR/PG_pg_dir = /TOOL_DIR/source/binPS_ps_dir = /TOOL_DIR/sourcePD_all_out = /out_directory/PA_all_fq_postfix = PostFix
三,运行命令
perl SOAPfuse-RUN.pl -c <config_file> -fd <WHOLE_SEQ-DATA_DIR> -l <sample_list> -o <out_directory> [Options]
运行的非常慢!!!
四,数据结果解读