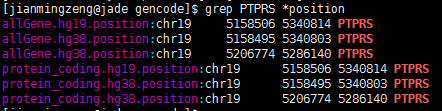
查了好久的bug,终于搞清楚问题所在了!因为要对基因进行reads计数,所以要拿到基因在基因组上面的染色体起始终止坐标,结果发现了个十分诡异的现象,很多基因有多个坐标,比如下面这个PTPRS 在hg38这个基因组版本,居然有两个定位,因为我是写程序格式化得到的坐标,所以我check了我的程序,http://www.biotrainee.com/thread-472-1-1.html 感兴趣的同学可以点开看看我的代码!
十一
10
查了好久的bug,终于搞清楚问题所在了!因为要对基因进行reads计数,所以要拿到基因在基因组上面的染色体起始终止坐标,结果发现了个十分诡异的现象,很多基因有多个坐标,比如下面这个PTPRS 在hg38这个基因组版本,居然有两个定位,因为我是写程序格式化得到的坐标,所以我check了我的程序,http://www.biotrainee.com/thread-472-1-1.html 感兴趣的同学可以点开看看我的代码!
最近跟一些志同道合的小伙伴们一起搭建了 生信技能树 的论坛,所以在社交上面加大了投入力度,认识了很多在生物信息学领域各个学习程度的同学,发现有些人问我的问题让哭笑不得,大意就是:我的生信菜鸟团博客有近四百篇文章,阅读10万+了,又是知乎上面的大V,现在又在建论坛,感觉生意很红火的样子,是不是挣了很多钱啊!
在许多小伙伴的共同协作下,我们的第一个论坛-生信技能树,诞生啦!
论坛地址:http://www.biotrainee.com/forum.php
虽然大家都说论坛已经是过气的互联网产品了,但我对互联网行业懂的很少,其实当初做博客的时候就有人跟我说过类似的话,但我还是坚持做了,我觉得做得还挺成功的,所以我仍然决定坚持把这个论坛做下去。
博客有很多缺点,传播速度很慢,不利于检索分类文章,个人知识面有限,也没办法跟follower及时交流。而我们的论坛,就可以克服那几个缺点。 Continue reading
估计很多小伙伴都没有听过sepsis,现在翻译成中文是脓毒病,很多人会把它与那个缺乏维他命C的败血症混淆,其实完全不一样,因为sepsis致死率非常高!
| sepsis | 英[ˈsepsɪs] | 美['sepsɪs] |
| n. | 脓毒病; 脓毒疾; |
自从我写了那篇关于创业的想法的文章后,传送门: 我也想开个公司(上) , 很多熟悉的朋友,还有不少陌生的朋友都给我来信,跟我讨论我的想法,尤其是几个海外的朋友特别热情,我们深度的讨论了创建自由职业者联盟的可行性,公司如何活下去,盈利点是什么,什么样的价值观才能铸就百年企业等各种话题,可能我们在创业这个领域都还是蹒跚学步的状态,但是大家的热心帮助让我很感动,这也坚定了我继续做知识传播者的想法。我这里简单分享一下我们讨论的几个公司战略方向,因为我还有四五年的博士生涯要度过,暂时无法全心全意的发展事业,如果有人看到了也想朝这个方向发展,我可以免费做咨询服务,我非常乐意看到有人能实现我的想法。 Continue reading
最近有很多朋友咨询我关于生物信息学数据处理的各种问题,有通过QQ直接对话聊天的,或者在QQ群里at我的,或者在知乎上面给我发短信息的,还有给我的163邮箱发信的。怎么说呢,首先还是感谢大家对我的信任,愿意花时间来跟我交流生物信息学数据处理的相关技术,然后我要简单说明一下为什么有些时候我没有答复你,虽然可能对你来讲,我是没有礼貌或者是太傲气了,但是我在这个领域浸淫了这么久,虽然你愿意跟我交流,但是你们的很多问题对我来说要么是都是太小儿科了,简单的google就能解决,要么是太空泛了,我无从答起,甚至我也给不出正确答案,更多的是有些人压根不用心的提问,纯粹是耽误你我的时间,所以我觉得很有必要写这篇博客简单说明一下,什么情况下我会回答你的问题。(如果你的问题非常吸引人,下面你就不用看了,我一定会抢着回答你的!) Continue reading
最近学习CHIP-seq的分析流程,略有点心得,也跟以前掌握的WES和RNA-seq做了一些比较,趁跑步的时候跟师妹讨论了一下,正好师妹写了一篇博客来分享这个讨论结果,我也借此机会转载过来,分享给大家,算是借花献佛吧!师妹的博文是用markdown写作,我觉得大家应该直接看她的文章,写得条理清楚:Exome-seq、ChIP-seq、RNA-seq之间的差异 Continue reading
呀,这是去年(2015)蹭的一个论坛,不记得是第几届了,反正是生物谷举办的,他们搞论坛已经成为一个产业了,非常挣钱的!我那时候还很认真的做了笔记,现在回过头来看看,他们好像讲的都很有道理,虽然我直到现在也用不上,不过我丝毫不担心。我一直拼命的学习各种知识,就是因为有着坚定的信念,所学的一切终将会有一天对我的人生有所帮助。
这也是对TCGA数据的深度挖掘,从而提出的一个统计学概念。文章研究了30种癌症,发现21种不同的mutation signature。如果理解了,就会发现这个其实蛮简单的,他们并不重新测序,只是拿已经有了的TCGA数据进行分析,而且居然是发表在nature上面!
研究了4,938,362 mutations from 7,042 cancers样本,突变频谱的概念只是针对于somatic 的mutation。一般是对癌症病人的肿瘤组织和癌旁组织配对测序,过滤得到的somatic mutation,一般一个样本也就几百个somatic 的mutation。
paper链接是:http://www.nature.com/nature/journal/v500/n7463/full/nature12477.html
在推特上面看到某人推荐了一篇好文,好像是某位大佬在science上面写的,大概就是鼓吹大家进入癌症研究领域。
http://science.sciencemag.org/content/352/6282/123.full
感觉写的还不错,就翻译了一下,译文如下:
我重新整理了一下我当初生信入门时候收集的资料,希望对后学者有帮助,如果你以前储存了我的分享,请删掉以前的, 重新转这个完整版,以后不会更新了,入门的资料其实很多都是一样的!
公开链接:http://pan.baidu.com/s/1jIvwRD8 ,没有密码,直接根据需求自行下载
因为视频教程架构出了点问题,所有不会更新了。现在公布所有下载链接,大家不需要去优酷观看了,太不清晰了,而且还有广告!
视频百度云盘链接:http://pan.baidu.com/s/1jIvwRD8
下面是以前写的大纲,虽然我不做视频了,但是大家可以按照下面的大纲来自学,希望能对你有所帮助!!!
我已经构建好bioconductor中文社区的雏形,大家可以进去看看!
突然发现我的bioconductor.cn这个域名都快要过期了!
哈哈,才想起一年前的计划到现在还没开始实施,实在不像我的风格,可能是水平到了一定程度吧,很多初级工作不像以前那样事无巨细的把关了。正好,借这个机会找几个朋友帮我一起完成这个bioconductor中文社区计划!
Hi,all. I just started to use github and found this magic way to write a blog.
so I do a simpler test here.
Don't be confused, there is nothing new for bioinformatic here.
And it very easy to use Markdown in wordpress,just download a plugin.
Below is some useful links to help you familar with markdown as soon as possible.
- http://bhttp://mahua.jser.me/log.csdn.net/kaitiren/article/details/38513715
- https://github.com/guodongxiaren/README/blob/master/README.md
- http://wowubuntu.com/markdown/
- http://mahua.jser.me/
Also I want to recommend some useful web-editor for markdown.
- http://mahua.jser.me/
- https://www.madoko.net/
- https://www.zybuluo.com/mdeditor
It's easy to use Markdown, but to get the most out of it, you still need to understand it and keep practising
因为大部分生物信息学软件都是linux版本的,所以生物信息学数据分析工作者必备技能就是linux,但是大部分人只是拿他当个中转站,我以前也是,直到接触了大批量的任务,自动化流程,才明白这里面的水太深了,不过无所谓,凭我个人的观点,其实shell的进阶语法真的不必要!
我也不想去搞明白操作符两边是否加空格的区别是什么了。
如果,还有其它功能需要实现,我们可以把脚本写的更负载,纯粹的用shell,需要搜索更多的shell技巧。
[perl]
## perl nohup.pl deep_count.sh 0
## perl nohup.pl deep_count.sh 1
## perl nohup.pl deep_count.sh 2
$i=1;
open FH,$ARGV[0];
while(<FH>){
chomp;
next unless $.%3==$ARGV[1];
$cmd="nohup $_ &";
print "$cmd\n";
system($cmd);
sleep(10800) if $i%5==4;
$i++;
#exit;
}
[/perl]
源码在这个package的github里面可以找到,有兴趣的童鞋可以研究一下
我们可以直接使用R的bioconductor里面的一个包,GOstats里面的函数来做超几何分布检验,看看每条pathway是否会富集
我们直接读取用limma包做好的差异分析结果
setwd("D:\\my_tutorial\\补\\用limma包对芯片数据做差异分析")
DEG=read.table("GSE63067.diffexp.NASH-normal.txt",stringsAsFactors = F)
View(DEG)
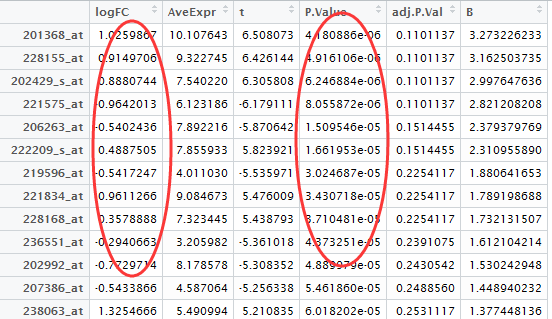
我们挑选logFC的绝对值大于0.5,并且P-value小雨0.05的基因作为差异基因,并且转换成entrezID
probeset=rownames(DEG[abs(DEG[,1])>0.5 & DEG[,4]<0.05,])
library(hgu133plus2.db)
library(annotate)
platformDB="hgu133plus2.db";
EGID <- as.numeric(lookUp(probeset, platformDB, "ENTREZID"))
length(unique(EGID))
#[1] 775
diff_gene_list <- unique(EGID)
这样我们的到来775个差异基因的一个list
首先我们直接使用R的bioconductor里面的一个包,GOstats里面的函数来做超几何分布检验,看看每条pathway是否会富集
library(GOstats)
library(org.Hs.eg.db)
#then do kegg pathway enrichment !
hyperG.params = new("KEGGHyperGParams", geneIds=diff_gene_list, universeGeneIds=NULL, annotation="org.Hs.eg.db",
categoryName="KEGG", pvalueCutoff=1, testDirection = "over")
KEGG.hyperG.results = hyperGTest(hyperG.params);
htmlReport(KEGG.hyperG.results, file="kegg.enrichment.html", summary.args=list("htmlLinks"=TRUE))
结果如下:
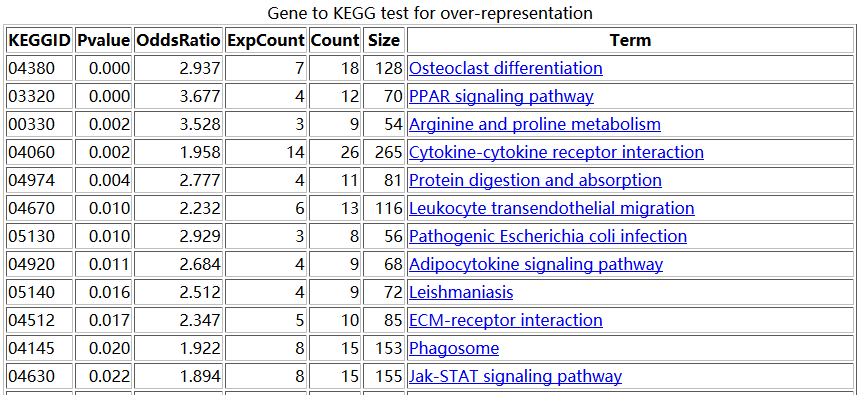
但是这样我们就忽略了其中的原理,我们不知道这些数据是如何算出来的,只是由别人写好的包得到了结果罢了。
事实上,这个包的这个hyperGTest函数无法就是包装了一个超几何分布检验而已。
如果我们了解了其中的统计学原理,我们完全可以写成一个自建的函数来实现同样的功能。
首先,我们要明白,limma接受的输入参数就是一个表达矩阵,而且是log后的表达矩阵(以2为底)。
那么最后计算得到的logFC这一列的值,其实就是输入的表达矩阵中case一组的平均表达量减去control一组的平均表达量的值,那么就会有正负之分,代表了case相当于control组来说,该基因是上调还是下调。
我之前总是有疑问,明明是case一组的平均表达量和control一组的平均表达量差值呀,跟log foldchange没有什么关系呀。
后来,我终于想通了,因为我们输入的是log后的表达矩阵,那么case一组的平均表达量和control一组的平均表达量都是log了的,那么它们的差值其实就是log的foldchange
首先,我们要理解foldchange的意义,如果case是平均表达量是8,control是2,那么foldchange就是4,logFC就是2咯
那么在limma包里面,输入的时候case的平均表达量被log后是3,control是1,那么差值是2,就是说logFC就是2。
这不是巧合,只是一个很简单的数学公式log(x/y)=log(x)-log(y)