我看到这篇science的补充材料最后一个图是: Continue reading
二
14
我看到这篇science的补充材料最后一个图是: Continue reading
高通量RNA测序已经能够更高效地检测融合转录本,但是融合检测的技术和相关软件通常产生高错误发现率。而一个自动整合RNA数据和已知基因组特征的可视化框架对于结果的检验是有帮助的。2017年发布的一个bioconductor包,chimeraviz就可以做到自动创建嵌合RNA可视化。
支持来自9种不同融合发现工具(deFuse、EricScript、InFusion、JAFFA、FusionCatcher、FusionMap、PRADA、SOAPfuse和STAR-FUSION)的输入。 Continue reading
该软件早在2016年就公布了,发表在biorxiv预印本上面,但直到2017年的双11,才发表在NG上面,文章是 : Annotation-free quantification of RNA splicing using LeafCutter 最大的特点应该是不需要参考基因组的基因注释信息了吧,就是gtf/gff文件可以省略,当然,比对还是需要的。它还有另外一个非常重要的功能,splicing quantitative trait loci (sQTLs) 但是跟我目前关系不大, 就不介绍了。 Continue reading
可变剪切是指mRNA前体以多种方式将exon连接在一起的过程。 由于可变剪切使一个基因产生多个mRNA转录本,不同mRNA可能翻译成不同蛋白。
转录组一般是指从细胞或组织的基因组所转录出来的RNA的总和,包括编码蛋白质的mRNA和各种非编码RNA(rRNA,tRNA,snRNA,snoRNA,lncRNA,microRNA等)。真核生物的基因结构是不连续的,如下图:
就是做一个图床而已,需要这个图片的网页url链接,没别的意思! Continue reading
其实这个植物是拟南芥,所以跟人类研究的数据处理大同小异。
转录组测序的研究对象为特定细胞在某一功能状态下所能转录出来的所有 RNA 的总和,包括 mRNA 和非编码 RNA 。通过转录组测序,能够全面获得物种特定组织或器官的转录本信息,从而进行转录本结构研究、变异研究、基因表达水平研究以及全新转录本发现等研究。 Continue reading
MeDIP-seq 跟ChIP-seq的分析手段是一模一样的,同理hMeDIP-seq,caMeDIP-seq等等,都没有本质上的区别,只是用的抗体不一样而已,请自行搜索基础知识,我只讲数据分析。
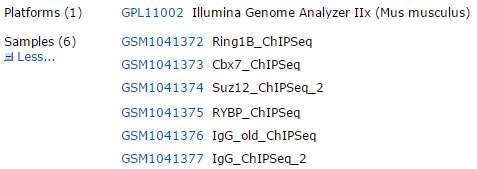

早在去年九月,我就写个博文说 RNA-seq流程需要进化啦! http://www.bio-info-trainee.com/1022.html ,主要就是进化成hisat2+stringtie+ballgown的流程,但是我一直没有系统性的讲这个流程,因为我觉真心木有用。我只用了里面的hisat来做比对而已!但是群里的小伙伴问得特别多,我还是勉为其难的写一个教程吧,你们之间拷贝我的代码就可以安装这些软件的!然后自己找一个测试数据,我的脚本很容易用的! Continue reading
我以前写过bedtools和htseq-counts的教程,它们都可以用来对比对好的bam文件进行计数,正好群里有小伙伴问我它们的区别,我就简单做了一个比较,大家可以先看看我以前写的软件教程。写的有的挫:
言归正传,我这里没精力去探究它们的具体原理,只是看看它们数一个read是否属于某个基因的时候,区别在哪里,大家看下图: Continue reading
表达芯片大家最熟悉的当然是affymetrix系列芯片啦,而且分析套路很简单,直接用R的affy包,就可以把cel文件经过RMA或者MAS5方法得到表达矩阵。illumina出厂的芯片略微有点不一样,它的原始数据有3个层级,一般拿到的是Processed data (示例), 当仍然需要一系列的统计学方法才能提取到表达矩阵。我比较喜欢用bioconductor,所以下面讲一讲如何用lumi包来处理这个芯片数据!
表达芯片大家最熟悉的当然是affymetrix系列芯片啦,而且分析套路很简单,直接用R的affy包,就可以把cel文件经过RMA或者MAS5方法得到表达矩阵。illumina出厂的芯片略微有点不一样,它的原始数据有3个层级,一般拿到的是Processed data (示例), 当仍然需要一系列的统计学方法才能提取到表达矩阵。接下来我们首先讲一讲illumina的bead 系列表达芯片基础知识吧: Continue reading