就是做一个图床而已,需要这个图片的网页url链接,没别的意思! Continue reading
十一
14
就是做一个图床而已,需要这个图片的网页url链接,没别的意思! Continue reading
使用annovar软件参考自:http://www.bio-info-trainee.com/?p=641
/home/jmzeng/bio-soft/annovar/convert2annovar.pl -format vcf4 Sample3.varscan.snp.vcf > Sample3.annovar
/home/jmzeng/bio-soft/annovar/convert2annovar.pl -format vcf4 Sample4.varscan.snp.vcf > Sample4.annovar
/home/jmzeng/bio-soft/annovar/convert2annovar.pl -format vcf4 Sample5.varscan.snp.vcf > Sample5.annovar
然后用下面这个脚本批量注释
Reading gene annotation from /home/jmzeng/bio-soft/annovar/humandb/hg19_refGene.txt ... Done with 50914 transcripts (including 11516 without coding sequence annotation) for 26271 unique genes
最后查看结果可知,真正在外显子上面的突变并不多
23515 Sample3.anno.exonic_variant_function
23913 Sample4.anno.exonic_variant_function
24009 Sample5.anno.exonic_variant_function
annovar软件就是把我们得到的十万多个snp分类了,看看这些snp分别是基因的哪些位置,是否引起蛋白突变
downstream
exonic
exonic;splicing
intergenic
intronic
ncRNA_exonic
ncRNA_intronic
ncRNA_splicing
ncRNA_UTR3
ncRNA_UTR5
splicing
upstream
upstream;downstream
UTR3
UTR5
UTR5;UTR3
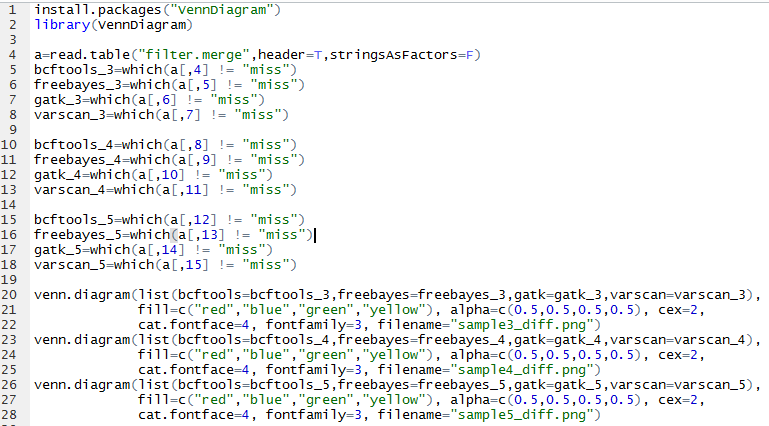
主要是画韦恩图看看,参考:http://www.bio-info-trainee.com/?p=893
对合并而且过滤的高质量snp信息来看看四种不同的snp calling软件的差异
我们用R语言来画韦恩图
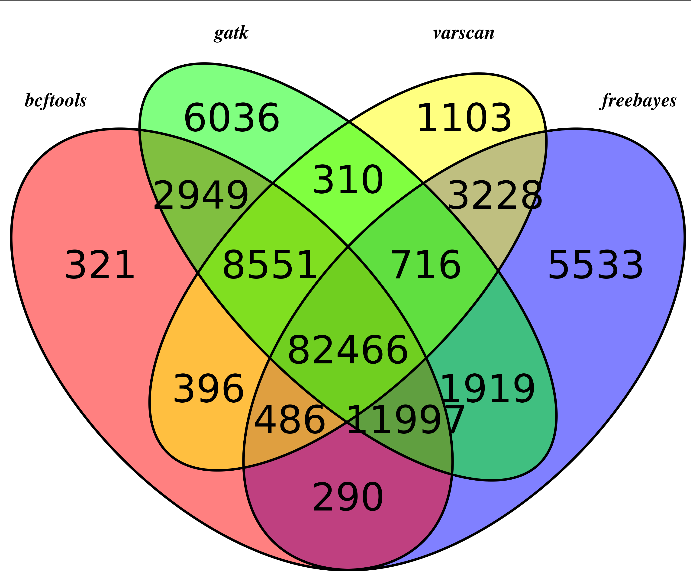
可以看出不同软件的差异还是蛮大的,所以我只选四个软件的公共snp来进行分析
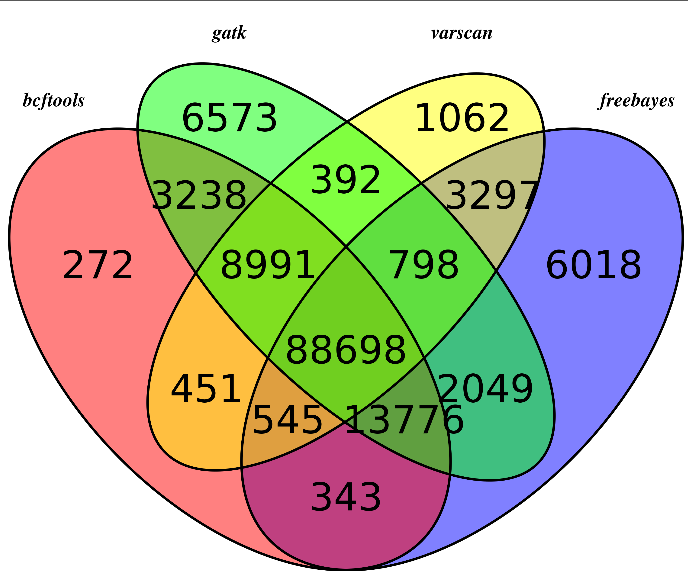
首先是sample3
然后是sample4
然后是sample5
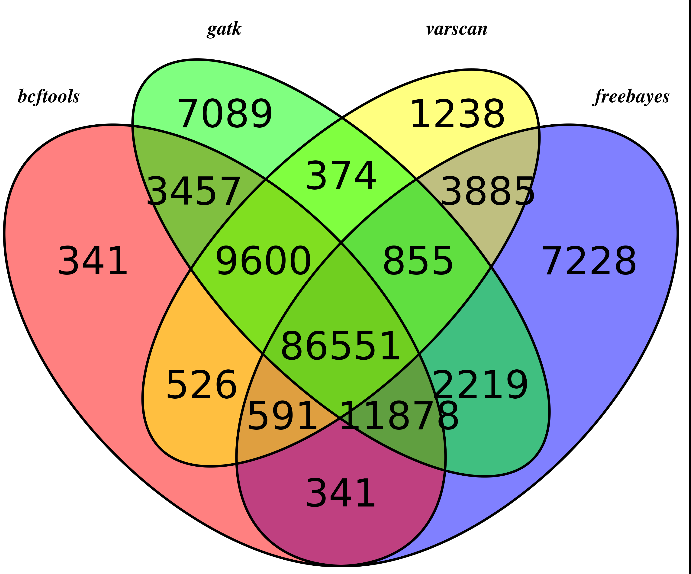
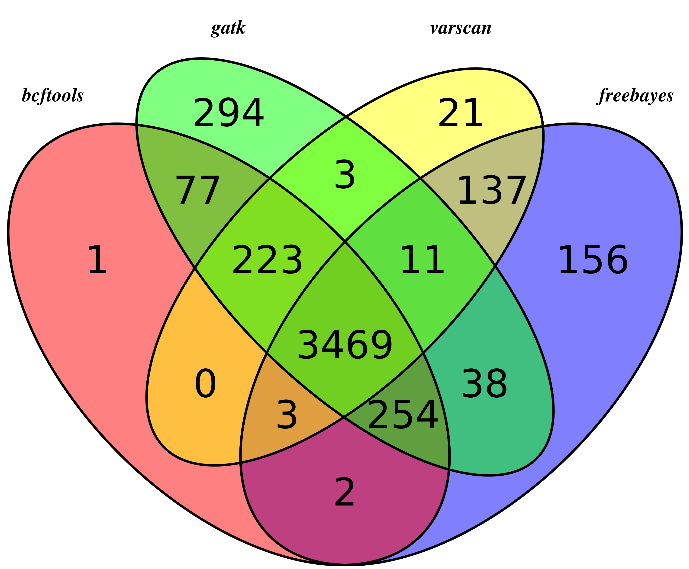
可以看出,不同的软件差异还是蛮大的,所以我重新比较了一下,这次只比较,它们不同的软件在exon位点上面的snp的差异,毕竟,我们这次是外显子测序,重点应该是外显子snp
然后我们用同样的程序,画韦恩图,这次能明显看出来了,大部分的snp位点都至少有两到三个软件支持
所以,只有测序深度达到一定级别,用什么软件来做snp-calling其实影响并不大。
3-4-5分别就是孩子、父亲、母亲
我对每个个体取他们的四种软件的公共snp来进行分析,并且只分析基因型,看看是否符合孟德尔遗传定律

结果如下:
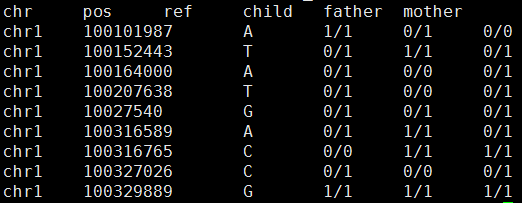
粗略看起来好像很少不符合孟德尔遗传定律耶
然后我写了程序计算

总共127138个可以计算的位点,共有18063个位点不符合孟德尔遗传定律,而且它们在染色体的分布情况如下
我检查了一下,不符合的原因,发现我把
chr1 100617887 C T:DP4=0,0,36,3 T:1/1:40 T:1/1:0,40:40 miss T:DP4=0,0,49,9 T:1/1:59 T:1/1:0,58:59 miss T:DP4=0,0,43,8 T:1/1:53 T:1/1:0,53:53 T:1/1:50
计算成了chr1 100617887 C 0/0 0/0 1/1 所以认为不符合,因为我认为只有四个软件都认为是snp的我才当作是snp的基因型,否则都是0/0
那么我就改写了程序,全部用gatk结果来计算。这次可以计算的snp有个176036,不符合的有20309,而且我看了不符合的snp的染色体分布,Y染色体有点异常
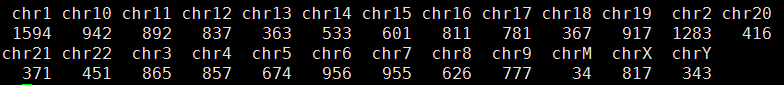
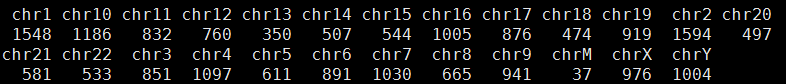
但是很失败,没什么发现!
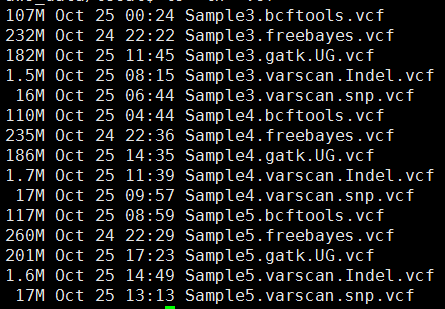
其中freebayes,bcftools,gatk都是把所有的snp细节都call出来了,可以看到下面这些软件的结果有的高达一百多万个snp,而一般文献都说外显子组测序可鉴定约8万个变异!
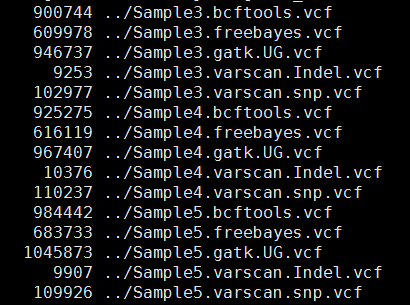
这样得到突变太多了,所以需要过滤。这里过滤的统一标准都是qual大于20,测序深度大于10。过滤之后的snp数量如下
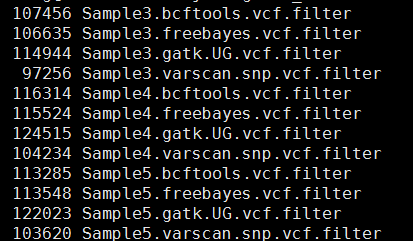
perl -alne '{next if $F[5]<20;/DP=(\d+)/;next if $1<10;next if /INDEL/;/(DP4=.*?);/;print "$F[0]\t$F[1]\t$F[3]\t$F[4]:$1"}' Sample3.bcftools.vcf >Sample3.bcftools.vcf.filter
perl -alne '{next if $F[5]<20;/DP=(\d+)/;next if $1<10;next if /INDEL/;/(DP4=.*?);/;print "$F[0]\t$F[1]\t$F[3]\t$F[4]:$1"}' Sample4.bcftools.vcf >Sample4.bcftools.vcf.filter
perl -alne '{next if $F[5]<20;/DP=(\d+)/;next if $1<10;next if /INDEL/;/(DP4=.*?);/;print "$F[0]\t$F[1]\t$F[3]\t$F[4]:$1"}' Sample5.bcftools.vcf >Sample5.bcftools.vcf.filter
perl -alne '{next if $F[5]<20;/DP=(\d+)/;next if $1<10;next unless /TYPE=snp/;@tmp=split/:/,$F[9];print "$F[0]\t$F[1]\t$F[3]\t$F[4]:$tmp[0]:$tmp[1]"}' Sample3.freebayes.vcf > Sample3.freebayes.vcf.filter
perl -alne '{next if $F[5]<20;/DP=(\d+)/;next if $1<10;next unless /TYPE=snp/;@tmp=split/:/,$F[9];print "$F[0]\t$F[1]\t$F[3]\t$F[4]:$tmp[0]:$tmp[1]"}' Sample4.freebayes.vcf > Sample4.freebayes.vcf.filter
perl -alne '{next if $F[5]<20;/DP=(\d+)/;next if $1<10;next unless /TYPE=snp/;@tmp=split/:/,$F[9];print "$F[0]\t$F[1]\t$F[3]\t$F[4]:$tmp[0]:$tmp[1]"}' Sample5.freebayes.vcf > Sample5.freebayes.vcf.filter
perl -alne '{next if $F[5]<20;/DP=(\d+)/;next if $1<10;next if length($F[3]) >1;next if length($F[4]) >1;@tmp=split/:/,$F[9];print "$F[0]\t$F[1]\t$F[3]\t$F[4]:$tmp[0]:$tmp[1]:$tmp[2]"}' Sample3.gatk.UG.vcf >Sample3.gatk.UG.vcf.filter
perl -alne '{next if $F[5]<20;/DP=(\d+)/;next if $1<10;next if length($F[3]) >1;next if length($F[4]) >1;@tmp=split/:/,$F[9];print "$F[0]\t$F[1]\t$F[3]\t$F[4]:$tmp[0]:$tmp[1]:$tmp[2]"}' Sample4.gatk.UG.vcf >Sample4.gatk.UG.vcf.filter
perl -alne '{next if $F[5]<20;/DP=(\d+)/;next if $1<10;next if length($F[3]) >1;next if length($F[4]) >1;@tmp=split/:/,$F[9];print "$F[0]\t$F[1]\t$F[3]\t$F[4]:$tmp[0]:$tmp[1]:$tmp[2]"}' Sample5.gatk.UG.vcf >Sample5.gatk.UG.vcf.filter
perl -alne '{@tmp=split/:/,$F[9];next if $tmp[3]<10;print "$F[0]\t$F[1]\t$F[3]\t$F[4]:$tmp[0]:$tmp[3]"}' Sample3.varscan.snp.vcf >Sample3.varscan.snp.vcf.filter
perl -alne '{@tmp=split/:/,$F[9];next if $tmp[3]<10;print "$F[0]\t$F[1]\t$F[3]\t$F[4]:$tmp[0]:$tmp[3]"}' Sample4.varscan.snp.vcf >Sample4.varscan.snp.vcf.filter
perl -alne '{@tmp=split/:/,$F[9];next if $tmp[3]<10;print "$F[0]\t$F[1]\t$F[3]\t$F[4]:$tmp[0]:$tmp[3]"}' Sample5.varscan.snp.vcf >Sample5.varscan.snp.vcf.filter
这样不同工具产生的snp记录数就比较整齐了,我们先比较四种不同工具的call snp的情况,然后再比较三个人的区别。
然后写了一个程序把所有的snp合并起来比较
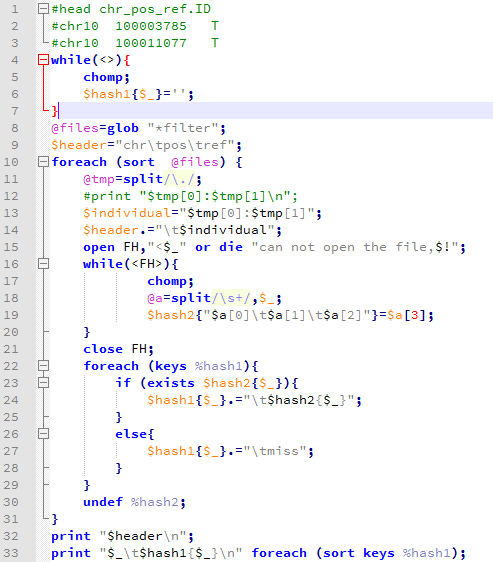
得到了一个很有趣的表格,我放在excel里面看了看 ,主要是要看生物学意义,但是我的生物学知识好多都忘了,得重新学习了 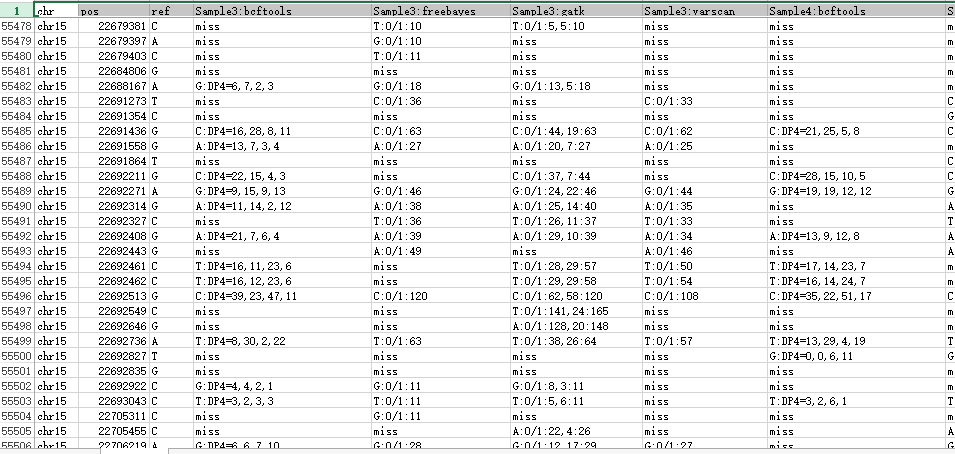
准备文件:下载必备的软件和参考基因组数据
1、软件
ps:还有samtools,freebayes和varscan软件,我以前下载过,这次就没有再弄了,但是下面会用到
2、参考基因组
3、参考 突变数据
第一步,下载数据
第二步,bwa比对
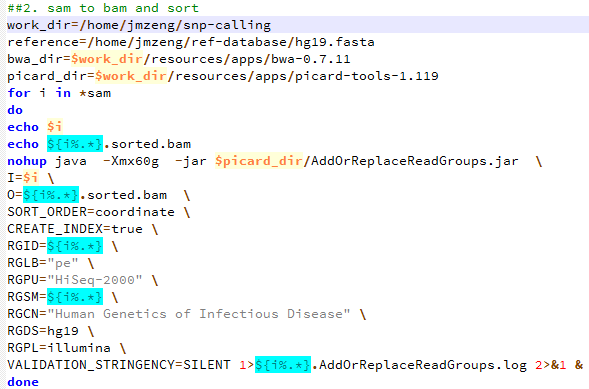
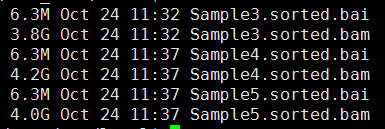
第三步,sam转为bam,并sort好
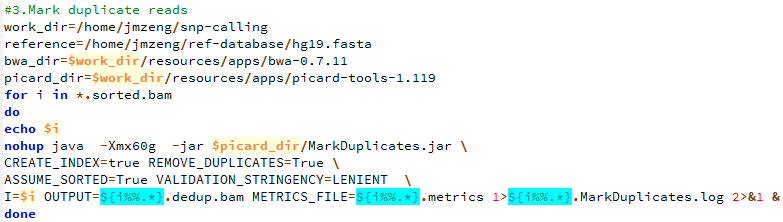
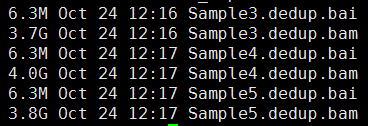
第四步,标记PCR重复,并去除
第五步,产生需要重排的坐标记录
第六步,根据重排记录文件把比对结果重新比对
第七步,把最终的bam文件转为mpileup文件
第八步,用bcftools 来call snp
第九步,用freebayes来call snp
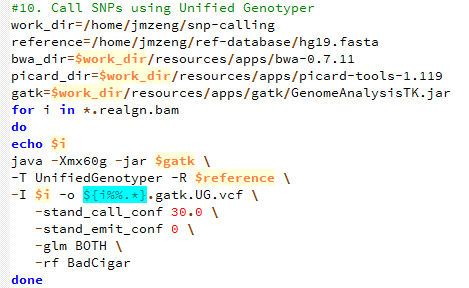
第十步,用gatk 来call snp
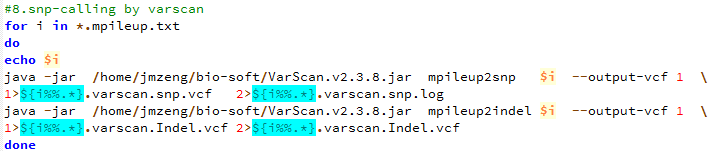
第十一步,用varscan来call snp
下面的图片是按照顺序来的,我就不整理了

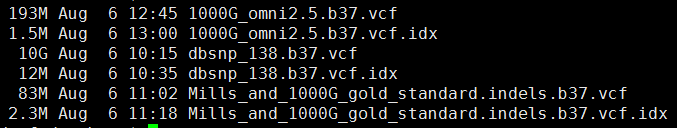
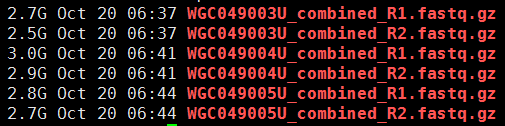
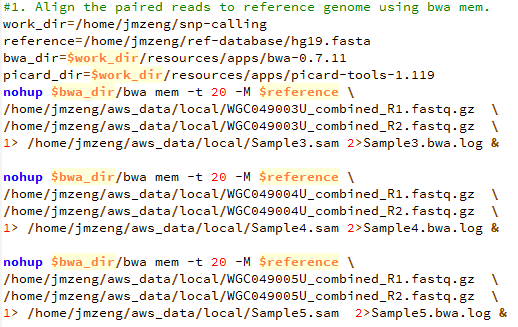
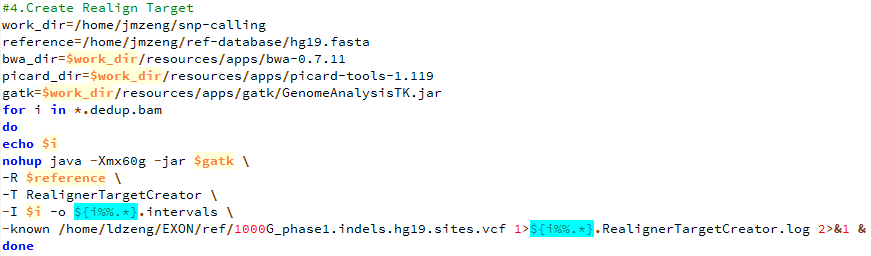
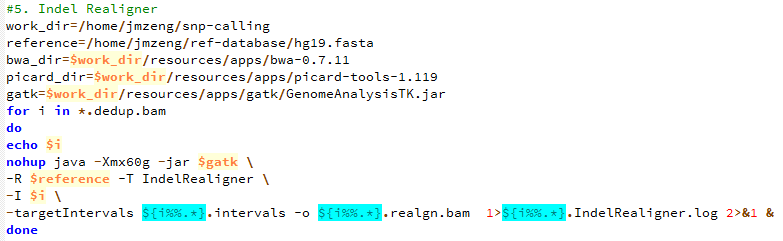
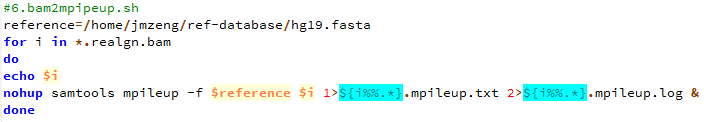
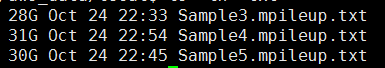
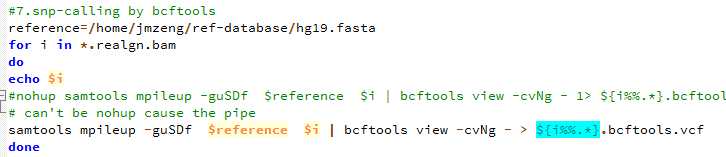
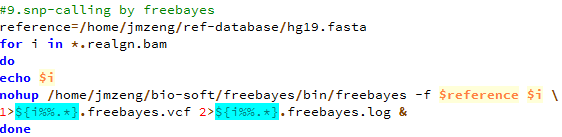
这一步主要看看这些外显子测序数据的测序质量如何: