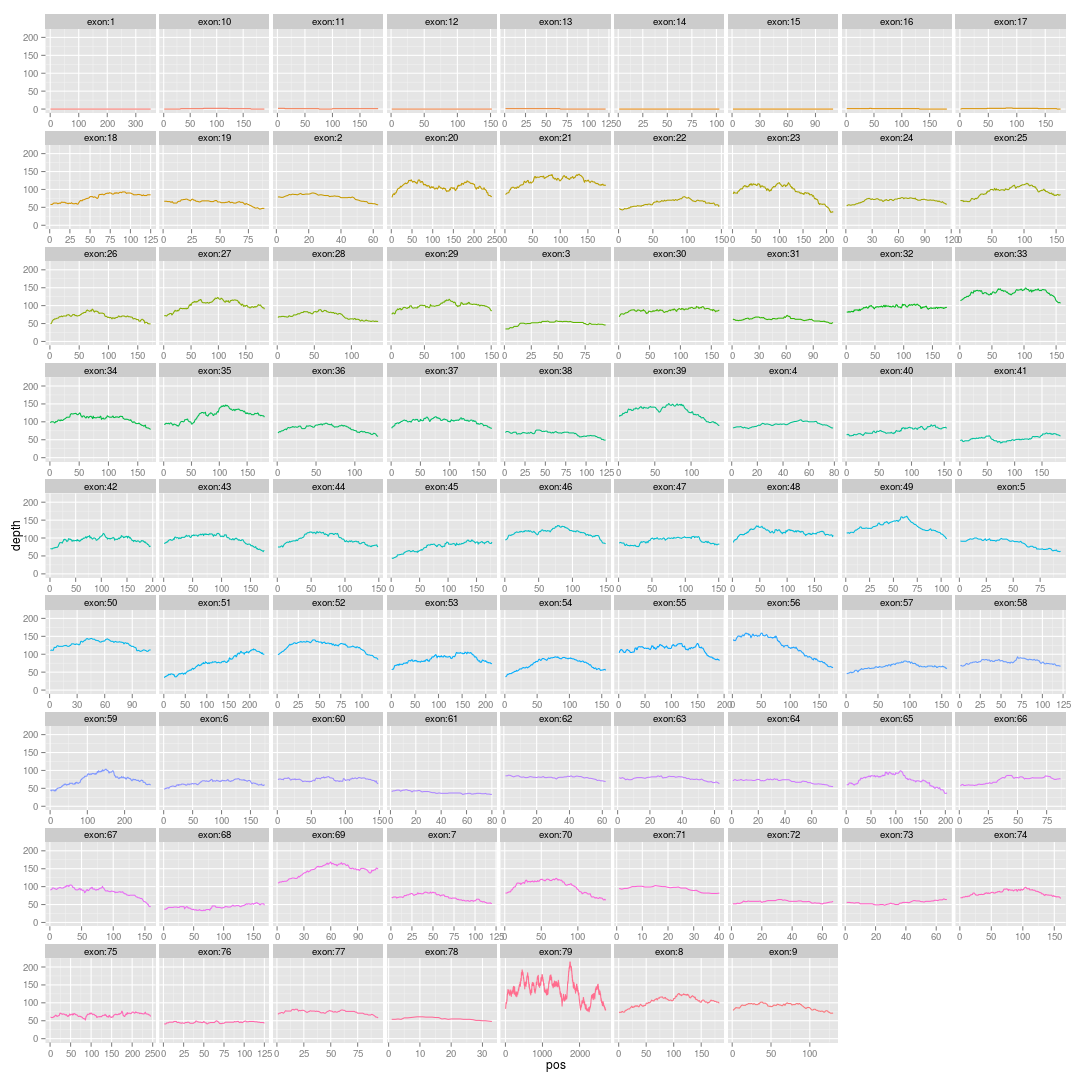
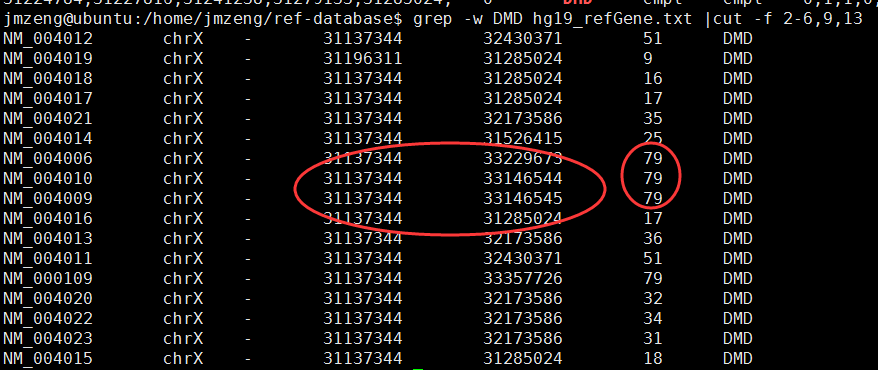
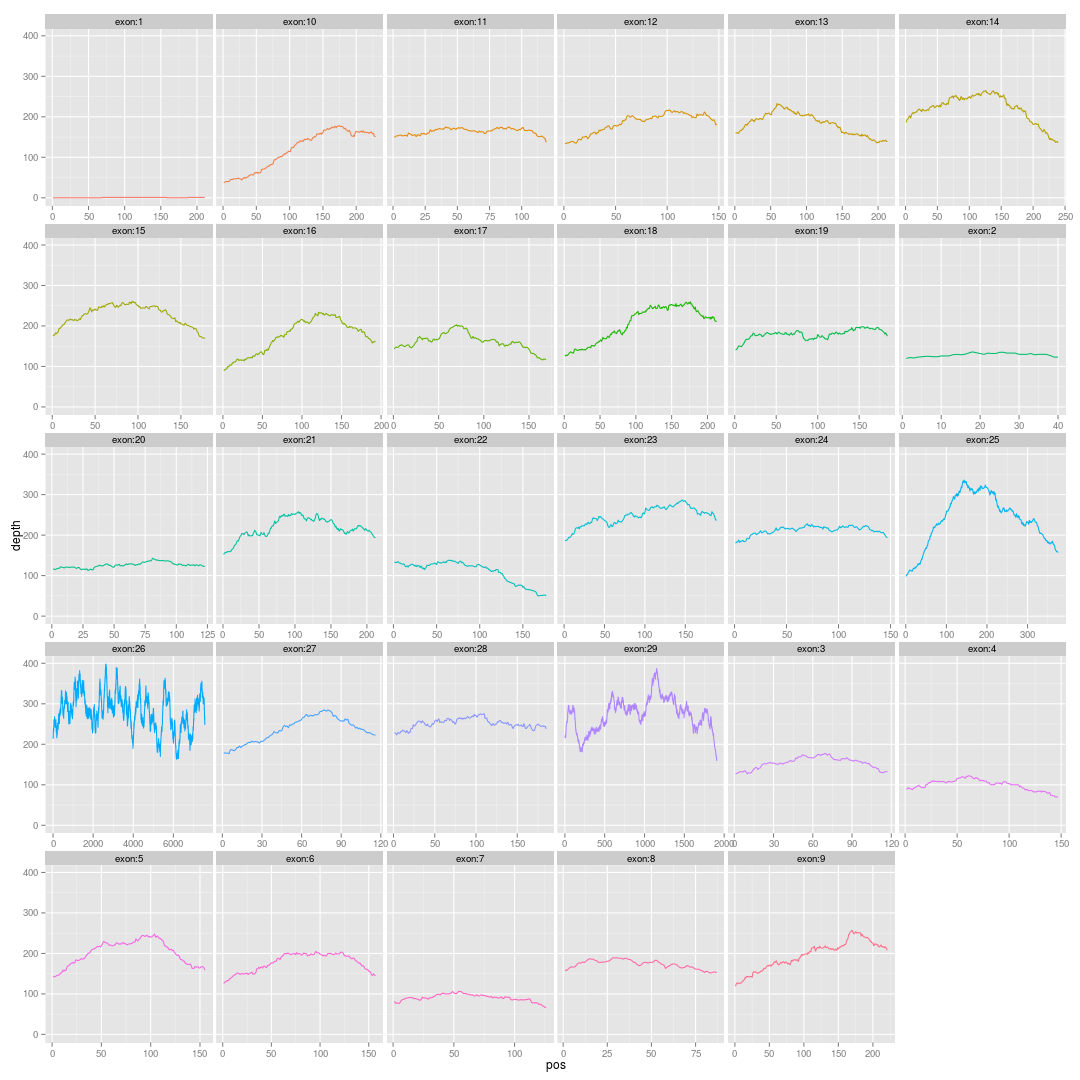
主要参考这篇文章的图4:http://www.nature.com/ng/journal/v42/n11/fig_tab/ng.691_F4.html Continue reading
九
02
该软件主页 http://bix.ucsd.edu/repeatscout/
wget http://bix.ucsd.edu/repeatscout/RepeatScout-1.0.5.tar.gz
解压进入目录,make即可
对于草莓这个215M的基因组来说,还是蛮快的!
第一步:用build_lmer_table命令把整个基因组生成一个频率表格,把所有有过重复的kmer都找出来。
/opt/RepeatScount/build_lmer_table -l 14 -sequence strawberry.fa -freq strawberry.freq
第二步:用 RepeatScout 这个命令根据生成的频率表格和基因组序列产生一个包含有所有的能找到的重复元件的文件。
RepeatScout -sequence strawberry.fa -freq strawberry.freq -l 14 -output strawberry_repeat
第三步:用filter-stage-1.prl这个脚本过滤掉低复杂度和串联重复元件。
貌似得到的文件为空,难道是全部过滤掉了???
第四步:需要借用repeatmasker来把这个得到repeat文件当作文库运行生成一个out文件
这个软件的参数其实蛮多的,我只是简单介绍了一些,关于它参数的调试,在我网盘里面还有更具体的文档说明,就不列了!
这是很久以前的一篇文章,我先贴出来给大家看看,然后讲一个实例
一:RepeatMasker的一些参数运行结果比较
从ncbi随便下载的zebrafish的一条sequence.fasta
不加上任何参数跑出来结果是 RepeatMasker sequence.fasta
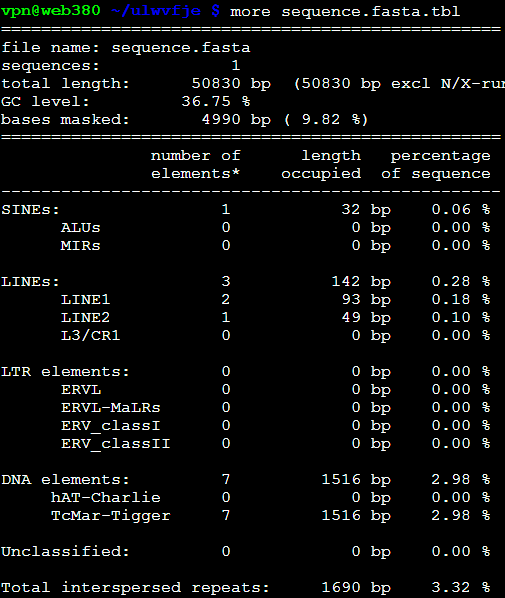
加上物种的参数之后跑出来是: RepeatMasker -species Danio sequence.fasta
效果里面出来了,之前得到的重复序列不到10%,这次可以达到70%以上,所以必须得选好对应的物种,这样才不会错过那么多要找的重复序列
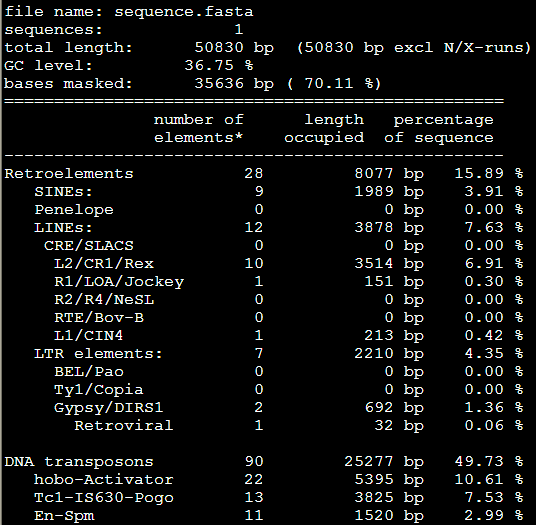
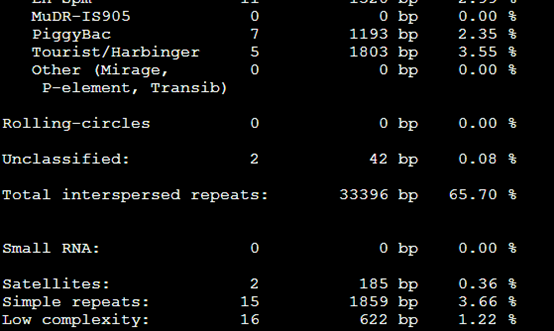
再加上-low这个参数是 RepeatMasker -species Danio -low sequence.fasta
感觉没有改变多少,就少了几个
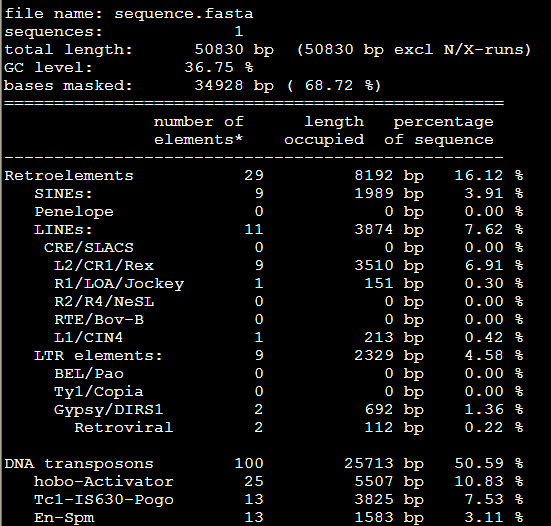
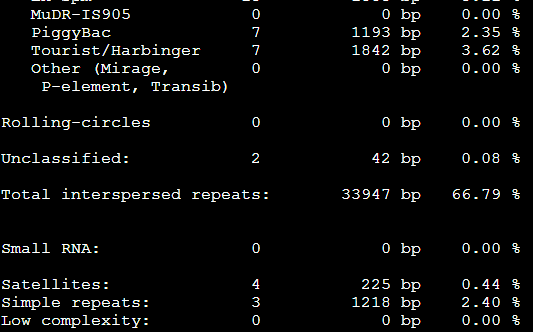
比较-div参数:RepeatMasker -species Danio sequence.fasta
RepeatMasker -species Danio -div 10 sequence.fasta
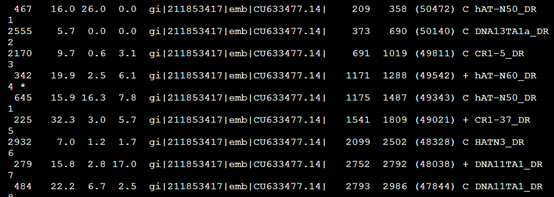
而加上-div 10之后
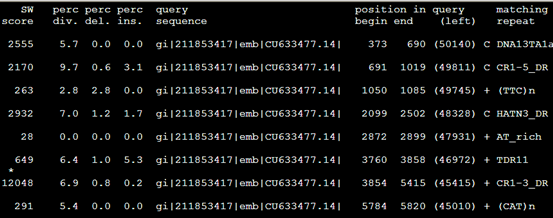
第二列小于10%的全部被剔除掉了
输出参数,本来应该是用N把重复区域屏蔽掉的
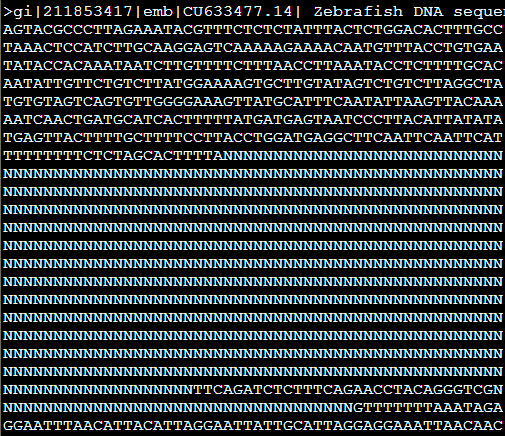
但是如果加上参数-x,原来输出是N的地方就都变成了X,感觉这个参数没啥子意义。
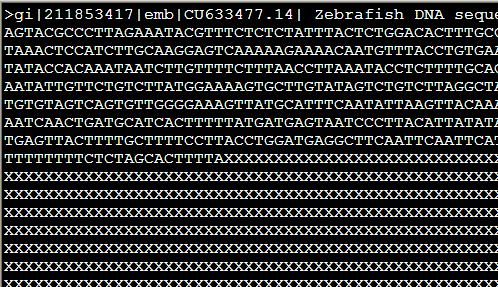
还有一些类似的参数,意义也不大,加上-xsmall,就是把重复区域用小写字母,不再需要N来掩盖了
如果加上-a这个参数,就多了一个文件
查看可知其内容是
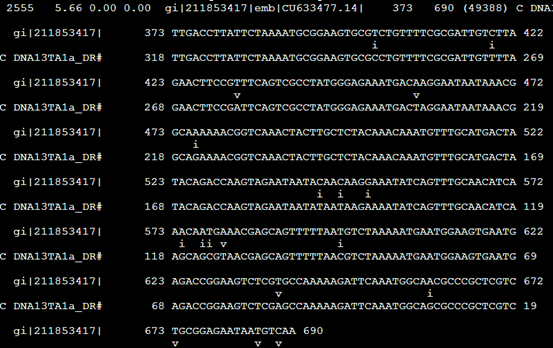
The alignments are in the cross_match/SWAT format, in which mismatches rather than matches are indicated: transitions
with an i and transversions with a v. Note it exists some differences between the alignment file and the map fi le.
The map fi le is produced by ProcessRepeats that the main task is to defragment the original map file and the alignment fi le is created from the original map fi le: the difference between them comes from the defragmented hits.
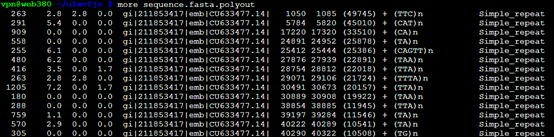
如果加上-poly,也会多出一个文件
![]()
查看,可知其单独列出了微卫星的表格
The ‘-xm’, ‘-ace,’ and ‘-gff ’ options create an additional out put file in cross match, ACeDB, and Gene Feature Finding format respectively.这几个参数都是为了生成适合其它处理的文件。
另外针对大文件的操作,可能需要-pa来设置运行速度,或者-s,-q,-qq
二:生成的文件的解释
会输出这些文件
![]()
1,。Out类文件
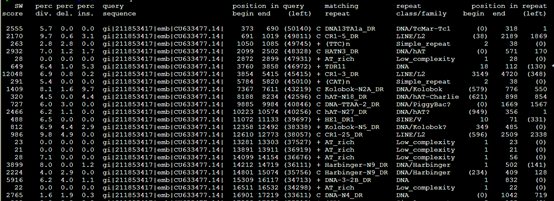
| SW score | 根据Smith-Waterman算法比对的分值 | 2555 | |
| Div% | 比上区间与共有序列相比的替代率 | 5.7 | |
| Del% | 在查询序列中碱基缺失的百分率(删除碱基) | 0.0 | |
| Ins% | 在repeat库序列中碱基缺失的百分率(插入碱基) | 0.0 | |
| Query sequence | 输入的待屏蔽重复的序列 | gi|211853417|emb|CU633477.14| | |
| Position begin | 373 | ||
| Position end | 690 | ||
| Query left | 在查询序列中超出比上区域的碱基数
+= 比上了库中重复序列的正义链,如果是互补连用“c”表示 |
(50140) | |
| Matching repeat | 比上的重复序列的名称 | C DNA13TA1a_DR | |
| Repeat family(class) | 比上的重复序列的类型 | DNA/TcMar-Tc1 | |
| Position begin | |||
| Position end | |||
| Query left | 比对区域距重复序列左端的碱基数 | ||
| 比对的顺序ID |
3.cat文件基本类似于。Out文件
3。。Tbl类文件
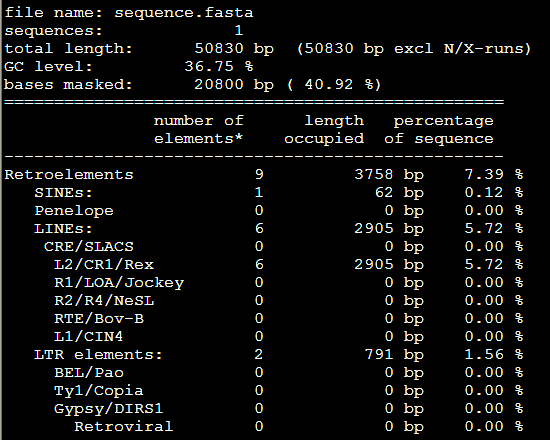
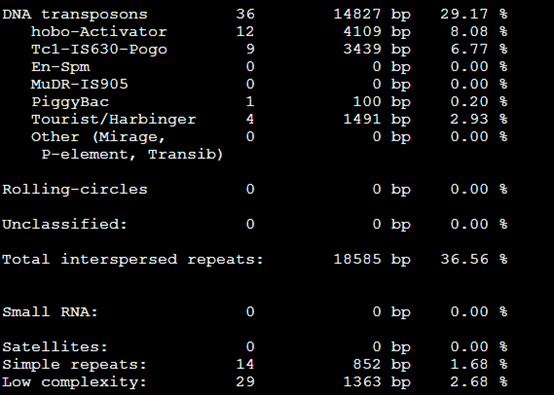
4.masked文件,就是找到的重复序列被N给代替了,或者用参数改变代替形式
polyout文件。就是单独列出了微卫星表格
Align文件,其实就是把之前的。Out文件的每一行记录单独拿出来再进行表格化解释
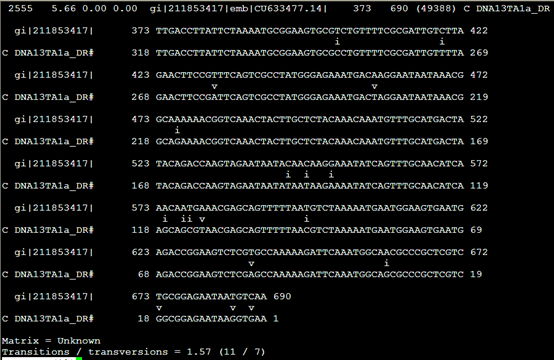
把373到690的核苷酸序列列出来,说明这个DNA13TA1a_DR 重复具体的意义
但是没看懂这个i,v是什么意思
结果比较
从ncbi随便下载的zebrafish的一条sequence.fasta
不加上任何参数跑出来结果是 RepeatMasker sequence.fasta
加上物种的参数之后跑出来是: RepeatMasker -species Danio sequence.fasta
效果里面出来了,之前得到的重复序列不到10%,这次可以达到70%以上,所以必须得选好对应的物种,这样才不会错过那么多要找的重复序列
这里我选取酵母基因组来组装,以为它只有一条染色体,而且本身也不大!
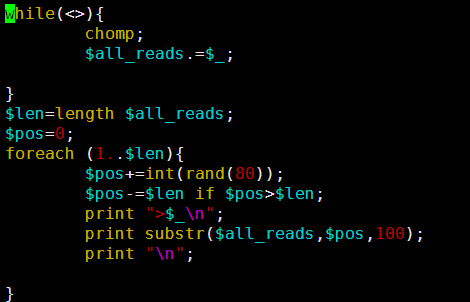
这个文件就4.5M,然后第一行就是序列名,第二列就是序列的碱基组成。共4641652个碱基。
我写一个perl程序来人为的创造一个测序文件
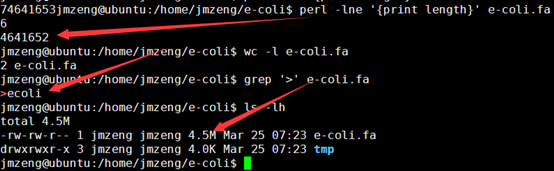
这样我们的4.5M基因组就模拟出来了486M的单端100bp的测序数据,而且是无缝连接,按照道理应该很容易就拼接的。
/home/jmzeng/bio-soft/SOAPdenovo2-bin-LINUX-generic-r240/SOAPdenovo-127mer
all -s config_file -K 63 -R -o graph_prefix 1>ass.log 2>ass.err
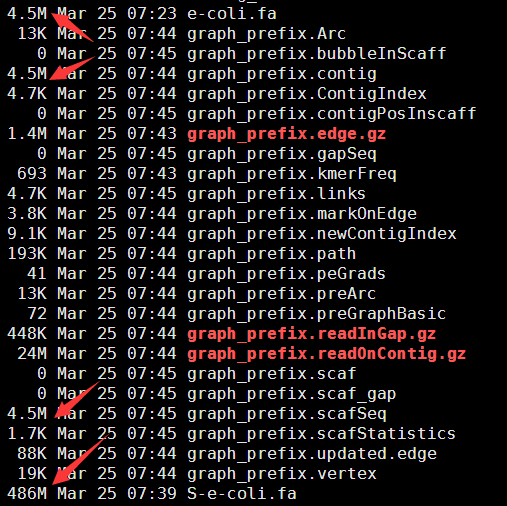
可以看到组装效果还不错哦,然后我模拟了一个测试数据,再进行组装一次,这次更好!
其实还可以模拟双端测序,应该就能达到百分百组装了。
但是由于我代码里面选取的是80在随机错开,所以我把kmer的长度设置成了81来试试看,希望这样可以把它完全组装成一条e-coli基因组。
/home/jmzeng/bio-soft/SOAPdenovo2-bin-LINUX-generic-r240/SOAPdenovo-127mer
all -s config_file -K 81 -R -o graph_prefix 1>ass.log 2>ass.err
但是也没有什么实质性的提高,虽然理论上是肯定可以组装到一起!
那我再模拟一个双端测序吧,中间间隔200bp的。
一.下载并安装这个软件
下载地址进下面,但是下载源码安装总是很困难,我直接下载bin文件可执行程序。
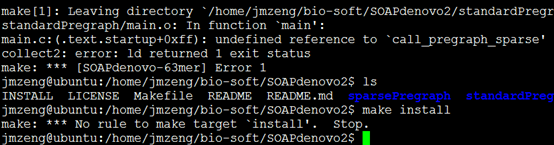
解压进入目录
首先make
然后make install即可
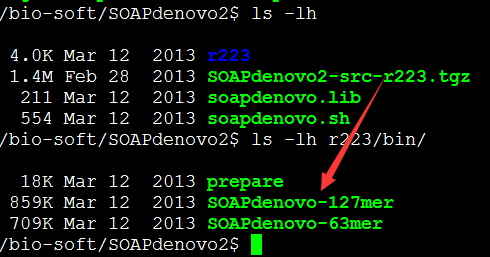
安装总是失败,我也不知道怎么回事,懒得解决了。
直接去我老师那里把这个程序拷贝进来了。
https://github.com/aquaskyline/SOAPdenovo2/archive/master.zip
http://sourceforge.net/projects/soapdenovo2/files/latest/download?source=files
也可以直接下载bin程序
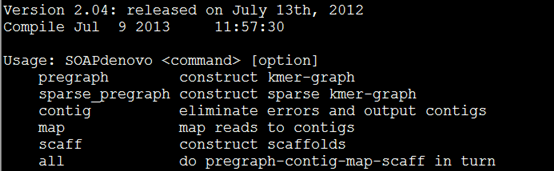
二.准备测试数据
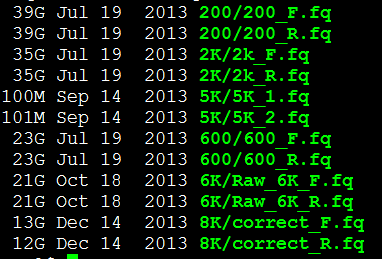
类似于这样的几个文库的左右两端测序数据。
我这里用一个小样本的单端数据做测试
三,参考命令
You may run it like this:
参考:http://www.plob.org/2012/07/06/2537.html
https://github.com/aquaskyline/SOAPdenovo2
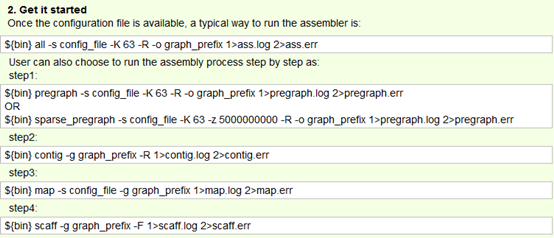
总共就四个步骤,介绍如下。
| ./pregraph_sparse [parameters] |
| ./SOAPdenovo-63mer contig [parameters] |
| ./SOAPdenovo-63mer map [parameters] |
| ./SOAPdenovo-63mer scaff [parameters] |
| i) preparing the pregraph. This step is similar to velveth for velvet. |
| ii) Determining contigs. This step is similar to velvetg for velvet. |
| iii) Mapping back reads on to contigs. |
| iv) Assembling contigs into scaffolds. |
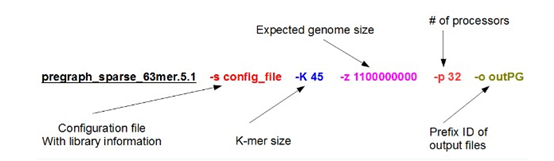
| SOAPdenovo-63mer sparse_pregraph -s config_file -K 45 -p 28 -z 1100000000 -o outPG |
| SOAPdenovo-63mer contig -g outPG |
| SOAPdenovo-63mer map -s config_file -g outPG -p 28 |
| SOAPdenovo-63mer scaff -g outPG -p 28 |
官网给出的步骤如下
这个命令还需要一个配置文件
max_rd_len=99 设置最大reads长度,具体情况具体定义
[LIB] 第一个文库数据
avg_ins=225
reverse_seq=0
asm_flags=3
rank=1
q1=runPE_1.fq
q2=runPE_2.fq
[LIB] 第二个文库数据
avg_ins=2000
reverse_seq=1
asm_flags=2
rank=2
q1=runMP_1.fq
q2=runMP_2.fq
也可以全部一次性的搞一个命令
all -s config_file -K 63 -R -o graph_prefix 1>ass.log 2>ass.err
我简单修改了一下参考博客的代码跟官网的代码,然后运行了我自己的代码
/home/jmzeng/bio-soft/SOAPdenovo2-bin-LINUX-generic-r240/SOAPdenovo-127mer
all -s config_file -K 63 -R -o graph_prefix 1>ass.log 2>ass.err
反正我也不懂,就先跑跑看咯
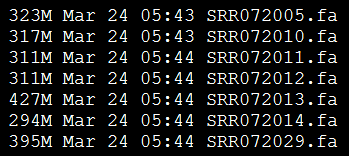
我选取的是7个单端数据,所以我的配置文件是
max_rd_len=500
[LIB]
avg_ins=225
reverse_seq=0
asm_flags=3
rank=1
p=SRR072005.fa
p=SRR072010.fa
p=SRR072011.fa
p=SRR072012.fa
p=SRR072013.fa
p=SRR072014.fa
p=SRR072029.fa
四.输出数据解读
好像我的数据都比较小,就7个三百多兆的fasta序列,几个小时就跑完啦
四个步骤都有输出数据
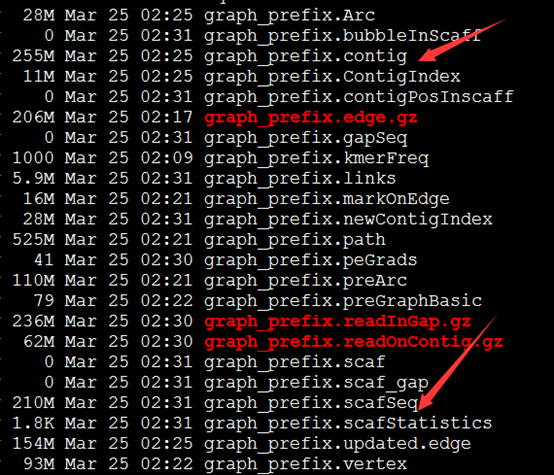
好像组装效果惨不忍睹呀!共86万的contig,50多万的scaffold
scaffolds>100 505473 99.60%
scaffolds>500 113523 22.37%
scaffolds>1K 48283 9.51%
scaffolds>10K 0 0.00%
scaffolds>100K 0 0.00%
scaffolds>1M 0 0.00%
这其实都相当于没有组装了,因为我的测序判断本来就很多是大于500的!
可能是我的kmer值选取的不对
Kmer为63跑出来的效果不怎么好,86万的contig,50万的scaffold的
Kmer为35跑出来的效果更惨,203万的contig,近60万的scaffold。
我觉得问题可能不是这里了,可能是没有用到那个20k和3k的双端测序库,唉,其实我习惯了illumina的测序数据,不太喜欢这个454的
感觉组装好难呀,业余时间搞不定呀,希望有高手能一起交流,哈哈,我自己再慢慢来试试。
今天先 对7个单端数据做处理,是454数据,平均长度300bp左右,明天再处理3KB和20KB的配对reads。
首先跑fastqc
打开一个个看结果
可以看到前面一些碱基的质量还是不错的, 因为这是454平台测序数据,序列片段长度差异很大,一般前四百个bp的碱基质量还是不错的,太长了的测序片段也不可靠
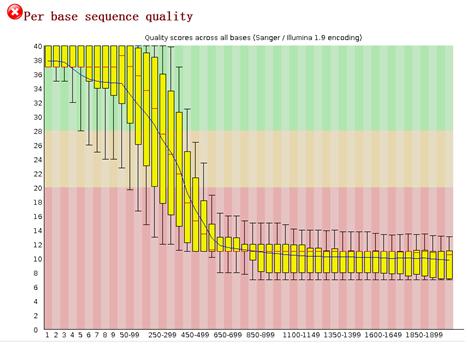
重点在下面这个图片,可以看到,前面的4个碱基是adaptor,肯定是要去除的,不是我们的测序数据。是TCAG,需要去除掉。
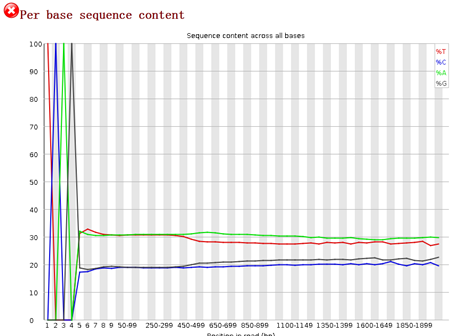
所以我们用了 solexaQA 这个套装软件对原始测序数据进行过滤
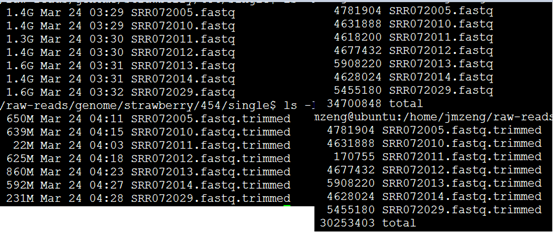
可以看到过滤的非常明显!!!甚至有个样本基本全军覆没了!然后我查看了我的批处理脚本,发现可能是perl DynamicTrim.pl -454 $id这个参数有问题
for id in *fastq
do
echo $id
perl DynamicTrim.pl -454 $id
done
for id in *trimmed
do
echo $id
perl LengthSort.pl $id
done
可以看到末尾的质量差的碱基都被去掉了,但是头部的TCAG还是没有去掉。
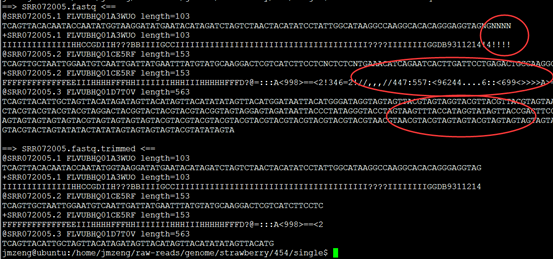
处理完毕后的数据如下:
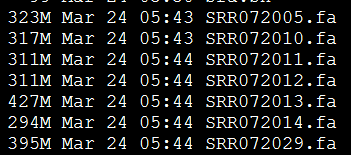
找橡胶测序数据无果
所以我只好找了他们所参考的草莓(strawberry, Fragaria vesca (2n = 2x = 14),a small genome (240 Mb),)的文章,是发表是nature genetics上面的
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3326587/
可以看到它的SRA索取号。

草莓组装结果:Over 3,200 scaffolds were assembled with an N50 of 1.3 Mb .
Over 95% (209.8 Mb) of the total sequence is represented in 272 scaffolds.
草莓基因息:Gene prediction modeling identified 34,809 genes, with most being supported by transcriptome mapping.
草莓染色体信息:Paradoxically, the small basic (x = 7) genome size of the strawberry genus, ~240 Mb,
offers substantial advantages for genomic research.
草莓来源:diploid strawberry F. vesca ssp. vesca accession Hawaii 4
(National Clonal Germplasm Repository accession # PI551572).
然后我去NCBI上面下载这三个数据
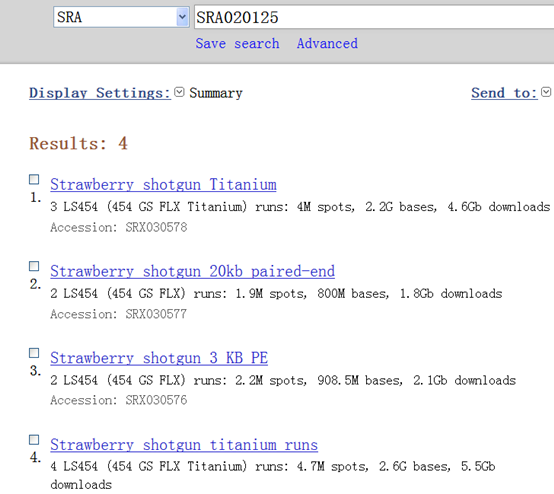
SRA020125 共有四个数据:
| http://www.ncbi.nlm.nih.gov/sra/SRX030575[accn] | Total: 4 runs, 4.7M spots, 2.6G bases, 5.5Gb |
| http://www.ncbi.nlm.nih.gov/sra/SRX030576[accn] (3 KB PE) | Total: 2 runs, 2.2M spots, 908.5M bases, 2.1Gb |
| http://www.ncbi.nlm.nih.gov/sra/SRX030577[accn] (20KB片段) | Total: 2 runs, 1.9M spots, 800M bases, 1.8Gb |
| http://www.ncbi.nlm.nih.gov/sra/SRX030578[accn] | Total: 3 runs, 4M spots, 2.2G bases, 4.6Gb |
挂在后台自动下载
![]()
好了,有了这些数据我们就要进行基因组的一系列分析啦!!!
不过我们可以先看看他们这个研究小组的成果
首先他们建造了一个关于草莓的基因组信息网站
https://strawberry.plantandfood.co.nz/
跟我之前在水科院做鲫鱼鲤鱼的差不多
直接在里面就可以下载他们做好的所有数据,也可以可视化。
它的染色体如下,非常简单,就七条染色体
http://www.rosaceae.org/species/fragaria/fragaria_vesca/genome_v1.1
我找到了它组装好的草莓基因组地址,用批处理全部下载了
