博文的顺序有点乱,因为怕读到前面的公共测序数据下载这篇文章的朋友搞不清楚,我如何调用各种软件的,所以我这里强势插入一篇博客来描述这件事,当然也只是略过,我所有的软件理论上都是安装在我的home目录下的biosoft文件夹,所以你看到我一般安装程序都是:
cd ~/biosoft
mkdir macs2 && cd macs2 ##指定的软件安装在指定文件夹里面 Continue reading
博文的顺序有点乱,因为怕读到前面的公共测序数据下载这篇文章的朋友搞不清楚,我如何调用各种软件的,所以我这里强势插入一篇博客来描述这件事,当然也只是略过,我所有的软件理论上都是安装在我的home目录下的biosoft文件夹,所以你看到我一般安装程序都是:
cd ~/biosoft
mkdir macs2 && cd macs2 ##指定的软件安装在指定文件夹里面 Continue reading
## step1 : download raw datacd ~mkdir CHIPseq_test && cd CHIPseq_testmkdir rawData && cd rawData## batch download the raw data by shell script :for ((i=593;i<601;i++)) ;do wget ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByStudy/sra/SRP/SRP033/SRP033492/SRR1042$i/SRR1042$i.sra;done
621M Jun 27 14:03 SRR1042593.sra (16.9M reads)2.2G Jun 27 15:58 SRR1042594.sra (60.6M reads)541M Jun 27 16:26 SRR1042595.sra (14.6M reads)2.4G Jun 27 18:24 SRR1042596.sra (65.9M reads)814M Jun 27 18:59 SRR1042597.sra (22.2M reads)2.1G Jun 27 20:30 SRR1042598.sra (58.1M reads)883M Jun 27 21:08 SRR1042599.sra (24.0M reads)2.8G Jun 28 11:53 SRR1042600.sra (76.4M reads)
## step2 : change sra data to fastq files.## cell line: MCF7 // Illumina HiSeq 2000 // 50bp // Single ends // phred+33ls *sra |while read id; do ~/biosoft/sratoolkit/sratoolkit.2.6.3-centos_linux64/bin/fastq-dump $id;donerm *sra
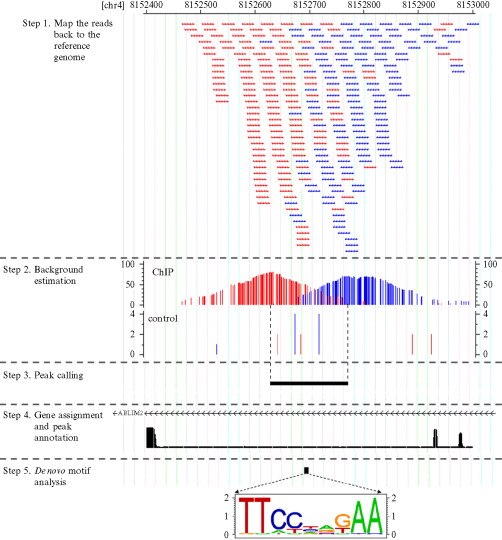
通过前面的分析,我们已经量化了ET1刺激前后的细胞的miRNA和mRNA表达水平,也通过成熟的统计学分析分别得到了差异miRNA和mRNA,这时候我们就需要换一个参考文献了,因为前面提到的那篇文章分析的不够细致,我这里选择了浙江大学的一篇TCGA数据挖掘分析文章Identifying miRNA/mRNA negative regulation pairs in colorectal cancer,里面首先就是查找miRNA-mRNA基因对,因为miRNA主要还是负向调控mRNA表达,所以根据我们得到的两个表达矩阵做相关性分析,很容易得到符合统计学意义的miRNA-mRNA基因对,具体分析内容如下:
把得到的差异miRNA的表达量画一个热图,看看它是否能显著的分类
用miRWalk2.0等数据库或者根据来获取这些差异miRNA的validated target genes
然后看看这些pairs of miRNA- target genes的表达量相关系数,选取显著正相关或者负相关的pairs
这些被选取的pairs of miRNA- target genes拿去做富集分析
最后这些pairs of miRNA- target genes做PPI网络分析
首先我们看第一个热图的实现:
resOrdered=na.omit(resOrdered)
DEmiRNA=resOrdered[abs(resOrdered$log2FoldChange)>log2(1.5) & resOrdered$padj <0.01 ,]
write.csv(resOrdered,"deseq2.results.csv",quote = F)
DEmiRNAexprSet=exprSet[rownames(DEmiRNA),]
write.csv(DEmiRNAexprSet,'DEmiRNAexprSet.csv')DEmiRNAexprSet=read.csv('DEmiRNAexprSet.csv',stringsAsFactors = F)
exprSet=as.matrix(DEmiRNAexprSet[,2:7])
rownames(exprSet)=rownames(DEmiRNAexprSet)
heatmap(exprSet)
gplots::heatmap.2(exprSet)
library(pheatmap)
## http://biit.cs.ut.ee/clustvis/
因为我前面保存的表达量就基于counts的,所以画热图还需要进行normalization,我这里懒得弄了,就用了一个网页版工具,自动出热图http://biit.cs.ut.ee/clustvis/
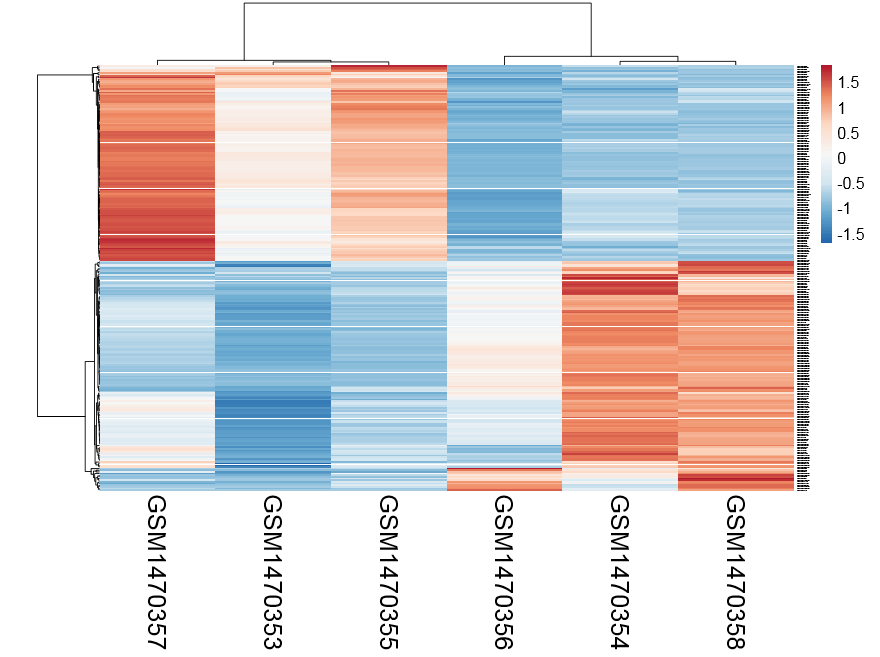
感觉还不错,可以很清楚的看到ET1刺激前后细胞中miRNA表达量变化
然后就是检验我们选取的感兴趣的有显著差异的miRNA的target genes,这时候有两种方法,一个是先由数据库得到已经被检验的miRNA的target genes,另一种是根据miRNA和mRNA表达量的相关性来预测。
用数据库来查找MiRNA的作用基因,非常多的工具,比较常用的有TargetScan/miRTarBase
### http://nar.oxfordjournals.org/content/early/2015/11/19/nar.gkv1258.full
### http://mirtarbase.mbc.nctu.edu.tw/
### http://mirtarbase.mbc.nctu.edu.tw/cache/download/6.1/hsa_MTI.xlsx
### http://www.targetscan.org/vert_71/ (version 7.1 (June 2016))
我还看到过一个整合工具: miRecords (DIANA-microT, MicroInspector, miRanda, MirTarget2, miTarget, NBmiRTar, PicTar, PITA, RNA22, RNAhybrid and TargetScan/TargertScanS)里面提到了查找MiRNA的作用基因这一过程,高假阳性,至少被5种工具支持,才算是真的
还有很多类似的工具,miRWalk2,psRNATarget网页版工具,最后值得一提的是中山大学的: starBase Pan-Cancer Analysis Platform is designed for deciphering Pan-Cancer Networks of lncRNAs, miRNAs, ceRNAs and RNA-binding proteins (RBPs) by mining clinical and expression profiles of 14 cancer types (>6000 samples) from The Cancer Genome Atlas (TCGA) Data Portal (all data available without limitations).虽然我没有仔细的用,但是看介绍好牛的样子,还有一个R包:miRLAB我玩了一会,它是先通过算所有配对的miRNA- genes的表达量相关系数,选取显著正相关或者负相关的pairs,然后反过来通过已知数据库来验证。
后面我就不讲了,主要看你得到miRNA的时候其它生物学数据是否充分,如果是癌症病人,有生存相关数据,可以做生存分析,如果你同时测了甲基化数据,可以做甲基化相关分析~~~~~~~~~
如果只是单纯的miRNA测序数据,可以回过头去研究一下de novo的miRNA预测的步骤,也是研究重点
这一讲其实算不上是自学miRNA-seq分析,本质就是affymetrix的mRNA表达芯片数据分析,而且还是最常用的那种GPL570 HG-U133_Plus_2,但是因为是跟miRNA样本配对检测的,而且后面会利用到这两个数据分析结果来做共表达网络分析等等,所以就贴出对该芯片数据的分析结果。文章里面也提到了 Messenger RNA expression analysis identified 731 probe sets with significant differential expression,作者挑选的差异分析结果的显著基因列表如下: Continue reading
这一讲是miRNA-seq数据分析的分水岭,前面的5讲说的是读文献下载数据比对然后计算表达量,属于常规的流程分析,一般在公司测序之后都可以拿到分析结果,或者文献也会给出下载结果。但是单纯的分析一个样本意义不大,一般来说,我们做研究都是针对于不同状态下的miRNA表达量差异分析,然后做注释,功能分析,网络分析,这才是重点,也是难点。我这里就直接拿文献处理好的miRNA表达量来展示如何做下游分析,首先就是差异分析啦: Continue reading
拿到比对后的sam/bam文件之后,这只能算是level2的数据,一般我们给他人share我们的结果也是直接给表达矩阵的, miRNA分析跟mRNA分析类似,但是它的表达矩阵更好获取一点。如果是mRNA,我们一般会跟基因组来比较,而基因组就那24条参考染色体,想知道具体比对到了哪个基因,需要根据基因组注释文件来写程序提取表达量信息,现在比较流行的是htseq这个软件,我前面也写过教程如何安装和使用,这里就不啰嗦了。但是对于miRNA,因为我比对的就是那1881条前体miRNA序列,所以直接分析比对的sam/bam文件就可以知道每条参考miRNA序列的表达量了。 Continue reading
序列比对是大多数类型数据分析的核心,如果要利用好测序数据,比对细节非常重要,我这里只是研读一篇文章也就没有对比对细节过多考虑,只是列出自己的代码和自己的几点思考,力求重现文章作者的分析结果。对miRNA-seq数据有两条比对策略,一种是下载miRBase数据库里面的已知miRNA序列来进行比对,一种直接比对到参考基因组(比如人类的是hg19/hg38),前面的比对非常简单,而且很容易就可以数出已经的所以miRNA序列的表达量,后面的比对有点耗时,而且算表达量的时候也不是很方便,但是它有个有点是可以来预测新的miRNA,所以大多数文章都会把这两条路给走一下。 Continue reading
因为我也是完全从零开始入门miRNA-seq分析,所以收集的资料比较齐全,我首先看了部分中文资料,了解了miRNA测序是怎么回事,该分析什么,然后主要围绕着上一篇提到的文献里面的分析步骤来搜索资料。传送门:自学miRNA-seq分析第一讲~文献选择与解
我首先拿到了miRNA定义:http://nar.oxfordjournals.org/content/34/suppl_1/D135.full ,当然基本上每个研究miRNA的文章都会在前言里面写到这个,我只是随意列出一个而已。 Continue reading
前些天逛bioStar论坛的时候看到了一个问题,是关于miRNA分析,提问者从NCBI的SRA数据下载文献提供的原始数据,然后处理的时候有些不懂,我看到他列出的数据是iron torrent测序仪的,而且我以前还没玩过miRNA-seq的数据分析, 就抽空自学了一下。因为我有RNA-seq的基础,所以理解学习起来比较简单。特记录一下自己的学习过程,希望对后学者有帮助。 Continue reading
我前面写到了生信分析人员如何入门linux和perl,后面还会写R和python的总结,但是在这中间我想插入一个脚本实战指南。其实在我前两篇日志里面也重点提到了学习编程语言最重要的就是实战了,也点出了几个关键词。在实际生物信息学数据处理中应用perl和linux,可以借鉴EMBOSS软件套件,fastx-toolkit等基础软件,实现并且模仿该软件的功能。尤其是SMS2/exonerate/里面的一些常见功能,还有DNA2.0 Bioinformatics Toolbox的一些工具。如果你这些名词不懂,请赶快谷歌!!! 它们做了什么,输入文件是什么,输出文件是什么,你都可以用脚本实现!
pwd/ls/cd/mv/rm/cp/mkdir/rmdir/man/locate/head/tail/less/morecut/paste/join/sort/uniq/wc/cat/diff/cmp/aliaswget/ssh/scp/curl/ftp/lftp/mysql/
软硬链接区别文本编辑,文件权限设置打包压缩解压操作(tar/gzip/bzip/ x-j x-c vf)软件的快捷方式如何实现?软件如何安装(源码软件,二进制可执行软件,perl/R/python/java软件)软件版本如何管理,各种编程语言环境如何管理,模块如何管理?(尤其是大部分没有root权限)
二是shell脚本,类似于windows的bat批处理文件
三是高级运维技巧
Hi,all. I just started to use github and found this magic way to write a blog.
so I do a simpler test here.
Don't be confused, there is nothing new for bioinformatic here.
And it very easy to use Markdown in wordpress,just download a plugin.
Below is some useful links to help you familar with markdown as soon as possible.
- http://bhttp://mahua.jser.me/log.csdn.net/kaitiren/article/details/38513715
- https://github.com/guodongxiaren/README/blob/master/README.md
- http://wowubuntu.com/markdown/
- http://mahua.jser.me/
Also I want to recommend some useful web-editor for markdown.
- http://mahua.jser.me/
- https://www.madoko.net/
- https://www.zybuluo.com/mdeditor
It's easy to use Markdown, but to get the most out of it, you still need to understand it and keep practising