我在生信技能树多次分享过生存分析的细节;
六
25
我在生信技能树多次分享过生存分析的细节;
最近在《生信技能树》公众号后台接到了求助,说他的肿瘤单细胞划分亚群的时候对T细胞的细分亚群把握不好,有超级简单的,也有非常复杂的,对于这样的求助,我也爱莫能助啊! Continue reading
前面我们开通了明码标价专栏:
不管是 GO或者KEGG这样的常见数据库的注释结果,还是mesh,reactomePA, DOSE这样的略微小众的数据库资源,不管是超几何分布检验的富集还是GSEA的算法,都Y叔都有对应的可视化函数支持。
众所周知,肿瘤的TNM分期是目前临床上比较常用的分期方式之一:
Continue reading
好的颜值,人人都爱,是你接触有趣的灵魂的敲门砖。单细胞数据分析也是如此,人人都知道需要降维聚类分群。 Continue reading
传统的bulk转录组测序并没有没落,虽然说大家都在抢单细胞的热点!
最近看到了一个胰腺癌的单细胞文章,公开了其测序数据及表达量矩阵,很方便做图表复现。 Continue reading
前面我们的明码标价之普通转录组上游分析,受到了各大热心粉丝的吐槽,觉得太简单了我们居然还好意思收费。后面我就就加上了稍微有一点难度的《可变剪切》,不过仍然是阻挡不了粉丝无穷无尽的需求,后台有人发给我一个RNA-Seq数据的内含子保留分析需求。
我看了看粉丝发过来的文章,发表于 January 2021, 在CELL杂志的文章《Spliceosome-targeted therapies trigger an antiviral immune response in triple-negative breast cancer》,链接是:https://doi.org/10.1016/j.cell.2020.12.031
这个文章数据比较多:
SUM159 SD6 RNA-Seq #GSE163414
LM2 SD6 RNA-Seq #GSE163411
SUM159 Cytoplasmic RNA-Seq #GSE163232
SUM159 J2 dsRIPseq #GSE163188
Syngeneic model RNA-Seq #GSE163181
可以看到,主要是RNA-Seq数据啦,有两个是普通的细胞系处理前后的表达量差异情况探索,所以出图如下:
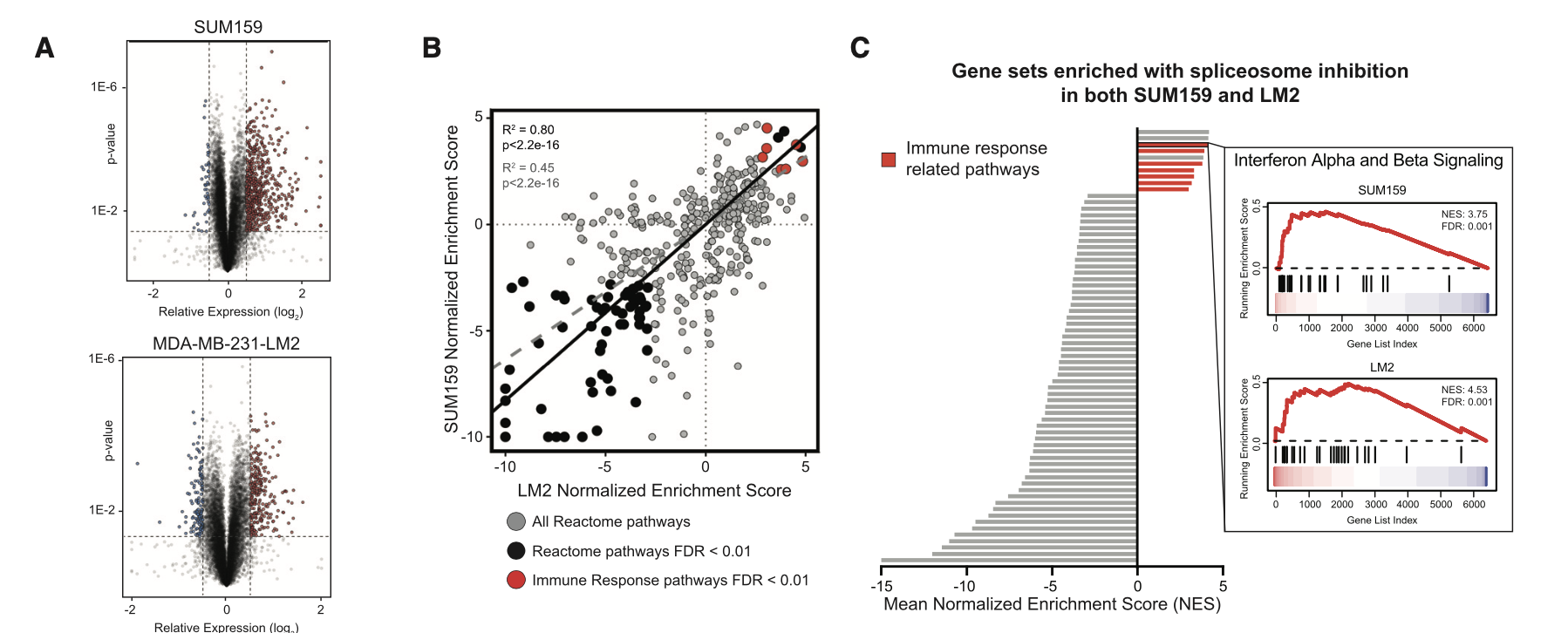
这个已经是超级简单了, 我们的明码标价之转录组常规测序服务(仅需799每个样品) 和 明码标价之普通转录组上游分析 就是对这样的 RNA-Seq拿到了表达量矩阵,然后下游分析也是平淡无奇,仅收费800,代码呢,我也多次分享了,基本上看我六年前的表达芯片的公共数据库挖掘系列推文即可;
这样的分析流程基本上绝大部分粉丝已经是无需委托我们啦,所以粉丝发给我的是 RNA-Seq数据的内含子保留分析需求,步骤如下:
对我们有ngs组学数据分析经验的人来说,其实并不难,无非就是安装几个软件,使用几个包。但对于没有学过编程的纯生物学研究者来说基本上不可能完成,也没有这样的网页工具。
但是呢,这个流程又确实是过于个性化,哪怕对我们来说很简单,也其实是耗费时间和精力需要研发调试的。
如果是TCGA数据库,步骤如下:
一般来说,大家是很难下载TCGA数据库原始fastq文件,这个权限审核比较严厉,不过咱们数据挖掘呢完全没有毕业一直盯着TCGA数据库啊,自己领域的普通RNA-seq肯定也是不少。如果是认真搞科研,你一定会自行调研和阅读文献,找到合适的数据集。
部分粉丝看到这里,可能无法理解RNA-Seq数据的内含子保留分析的意义是什么?
其实就是多了一个维度的指标,来把你的样本分类,分类后就可以找差异。同样的我们可以看这个示例文章,感觉每个样品的IR指标,把病人分成IR高低两个组别,然后走普通的ssGSEA分析,生存分析。
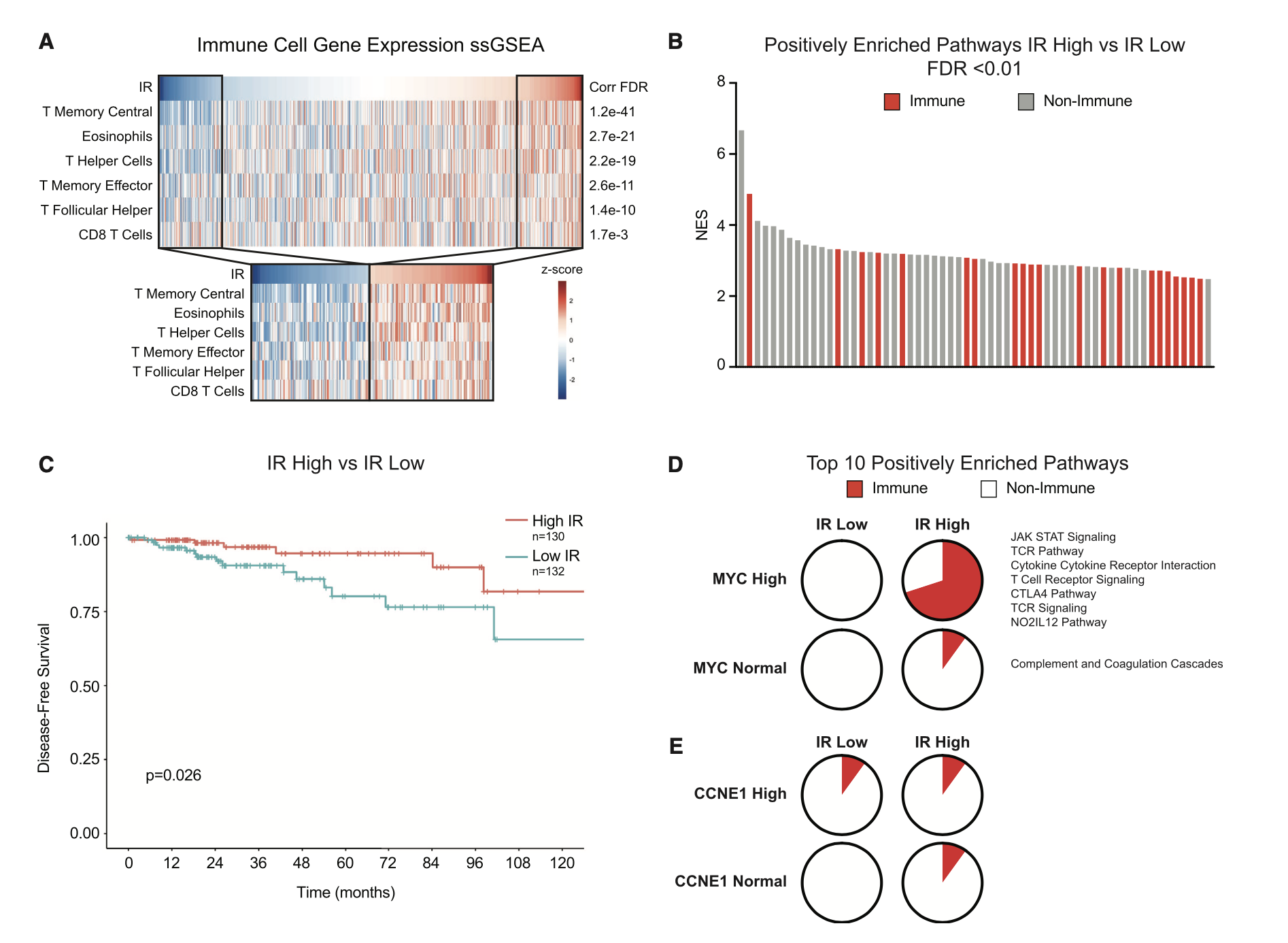
这一套组合拳,大家是不是很眼熟啊?
如果你也想做自己的的RNA-Seq数据的内含子保留分析,赶快联系我们吧,同样的,我们的分析仍然是明码标价,单个RNA-Seq数据的内含子保留分析收费仅需800元,因为是纯粹的基于Linux平台的各种软件脚本,所以提供你全套数据和脚本但是无法保证你能运行成功,因为你不一定有自己的服务器。
最近看到了朋友圈疯转的:[发paper怎么少走弯路?顶刊编辑手把手指点!【文字版】] Continue reading
我在:借鉴escape包的一些可视化GSVA或者ssGSEA结果矩阵的方法 和 对单细胞表达矩阵做gsea分析的两个教程里面提到过,MSigDB(Molecular Signatures Database)数据库中定义了已知的基因集合:http://software.broadinstitute.org/gsea/msigdb 需要注册才能下载。
但是这个GitHub包,ncborcherding/escape文档,在:http://www.bioconductor.org/packages/release/bioc/vignettes/escape/inst/doc/vignette.html 提供了一个封装好的MSigDB数据库信息,其实你仔细看它的文档,它的打包其实是依赖于msigdbr_7.2.1。
MigDB中的全部基因集 都被这个GitHub包,ncborcherding/escape 打包起来了,MSigDB(Molecular Signatures Database)数据库中定义了已知的基因集合:http://software.broadinstitute.org/gsea/msigdb 包括H和C1-C7八个系列(Collection),每个系列分别是:
GS <- getGeneSets(library = "H")
GS
MigDB中的全部基因集 被 构建成为: a list of GSEABase GeneSet objects ,获取 hallmark gene sets (癌症)特征基因集合。
安装方法非常简单:
install.packages("msigdbr")
但是这个msigdbr并没有我想象中的那么大:
Installing package into ‘C:/Users/win10/Documents/R/win-library/4.0’
(as ‘lib’ is unspecified)
试开URL’https://cran.rstudio.com/bin/windows/contrib/4.0/msigdbr_7.2.1.zip'
Content type 'application/zip' length 6737651 bytes (6.4 MB)
downloaded 6.4 MB
package ‘msigdbr’ successfully unpacked and MD5 sums checked
同样的,学习R包,看看文档即可,在: https://cran.r-project.org/web/packages/msigdbr/vignettes/msigdbr-intro.html
Documentation for package ‘msigdbr’ version 7.2.1
DESCRIPTION file.
User guides, package vignettes and other documentation.
Help Pages
msigdbr Retrieve the gene sets data frame
msigdbr_collections List the collections available in the msigdbr package
msigdbr_show_species List the species available in the msigdbr package
msigdbr_species List the species available in the msigdbr package
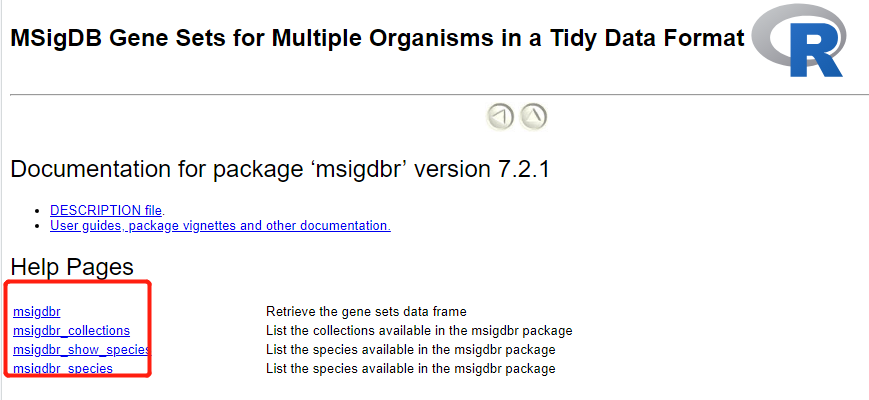
这些代码使用就明白了,确实没啥好继续讲解的:
library(msigdbr)
# All gene sets in the database can be retrieved without specifying a collection/category.
all_gene_sets = msigdbr(species = "Mus musculus")
head(all_gene_sets)
msigdbr_species()
all_gene_sets = msigdbr(species = "Homo sapiens")
无非就是封装和对象,前面的 escape 包提供了getGeneSets函数,我们的这个msigdbr提供了 msigdbr函数。
B站的10个小时教学视频务必看完,参考 GitHub 仓库存放的相关学习路线指导资料:https://github.com/jmzeng1314/R_bilibili ,可以参考一些优秀笔记,比如https://mubu.com/doc/2KUiSCfVsg
前面我们分享了 跟着Nature Medicine学MeDIP-seq数据分析,数据和代码都是公开,这个2G的压缩包文件,足以学习3个月,写60篇教程。 Continue reading
我们《生信技能树》早期也分享过蛋白质组学数据处理教程,目录如下:
Continue reading
GTEx数据库想必大家并不陌生了,通常我们在挖掘TCGA数据库的时候,会发现该项目纳入的正常组织测序结果是非常少的,也就是说很多病人都不会有他的正常组织的转录组测序结果。 Continue reading
四年前我做了一个单细胞课程,就是对scRNAseq包里面的数据示例的一些处理。 Continue reading
如果你看了我的单细胞转录组数据分析的 基础10讲:

做单细胞数据分析,我们当然希望看到一个清晰的降维聚类分群结果,这样才方便做生物学亚群注释,比如前面的例子:[人人都能学会的单细胞聚类分群注释] Continue reading
看到了发表于2020年8月的一个研究,标题是《Efficient chromatin profiling of H3K4me3 modification in cotton using CUT&Tag》,链接是 https://link.springer.com/article/10.1186/s13007-020-00664-8 Continue reading
众所周知,发布在bioconductor的包主要是生物信息学相关,在官方可以看到其主要是分成3类: Continue reading
绝大部分的肿瘤研究单细胞研究我介绍过 [CNS图表复现08—肿瘤单细胞数据第一次分群通用规则] Continue reading