昨天提到了最近接了一个单细胞转录组项目,有80个10X样品,每个样品的单细胞测序数据都是100G左右的fq.gz文件,在跑完了cellranger流程后整理结果的同时,重新捡起来了七八年前的Linux知识,写了个 带着文件夹结构的拷贝笔记分享给大家。 Continue reading
六
24
昨天提到了最近接了一个单细胞转录组项目,有80个10X样品,每个样品的单细胞测序数据都是100G左右的fq.gz文件,在跑完了cellranger流程后整理结果的同时,重新捡起来了七八年前的Linux知识,写了个 带着文件夹结构的拷贝笔记分享给大家。 Continue reading
正在发愁今天公众号写什么,电脑挂机的微信就看到了素未谋面的“学徒”负责的公众号《 医学生之学习生信 》跳出来了组学系列教程,好奇之下就点进去想看看他们更新到哪里了,一看吓一跳! Continue reading
我们的《生信入门》课程已经有两个年头了,培养了好几千学员,也有少部分极优秀的小伙伴有了自己的博客和公众号。
我注意到3个月的一个学员超级优秀,把我们的一个月授课内容系统性的整理成为了200篇左右的学习笔记,创立了一个公众号,精选目录如下:
Continue reading
前面我们开始接数据分析业务了,比如 明码标价之普通转录组上游分析,马上就在《生信技能树》公众号后台接到了很多需求。
最近有粉丝在我们《生信技能树》公众号后台付费求助,想follow一个文章看两个基因组合起来在一个数据集的生存分析。 Continue reading
最近读文献, 看到了一个有意思的文章,发表在 Nat Commun . 2021 Jan 的文章:《Global computational alignment of tumor and cell line transcriptional profiles 》提到了一个工具,Cellinger,链接是:https://www.nature.com/articles/s41467-020-20294-x
Continue reading
看到了朋友圈有人转发了这样的一个系列:
创作者的心声很有意思:想和大家一起开始学习生物信息学。因为最近看文献(涉及生信),总是大概明白了,自己还是无法操作。我也关注了很多生信的公众号,加了几个群,心动种草了一些线上课程。但是实在是觉得自己的基础知识太薄弱了,都看不懂群里的提问。所以决定要系统学一下,先看几本书吧,然后再学个线上操作课。
让我想起来了一个故事,就是现阶段生物信息学本科培养的人才与社会工作实践脱钩的问题。因为我看到他这个学习内容完全就是现阶段生物信息学本科培养大纲。
比如,他最新的一个是:【陪你学·生信】十二、RNA相关的简单分析,提到了Mfold 可以做RNA的二级结构预测,很有意思。在其网页工具上面输入你需要fold的序列,点击 fold RNA。如果事先不知道关于这段序列的任何信息,那么其他的参数都保持默认,就可以拿到该RNA序列的二级结构,确实是超级好用。
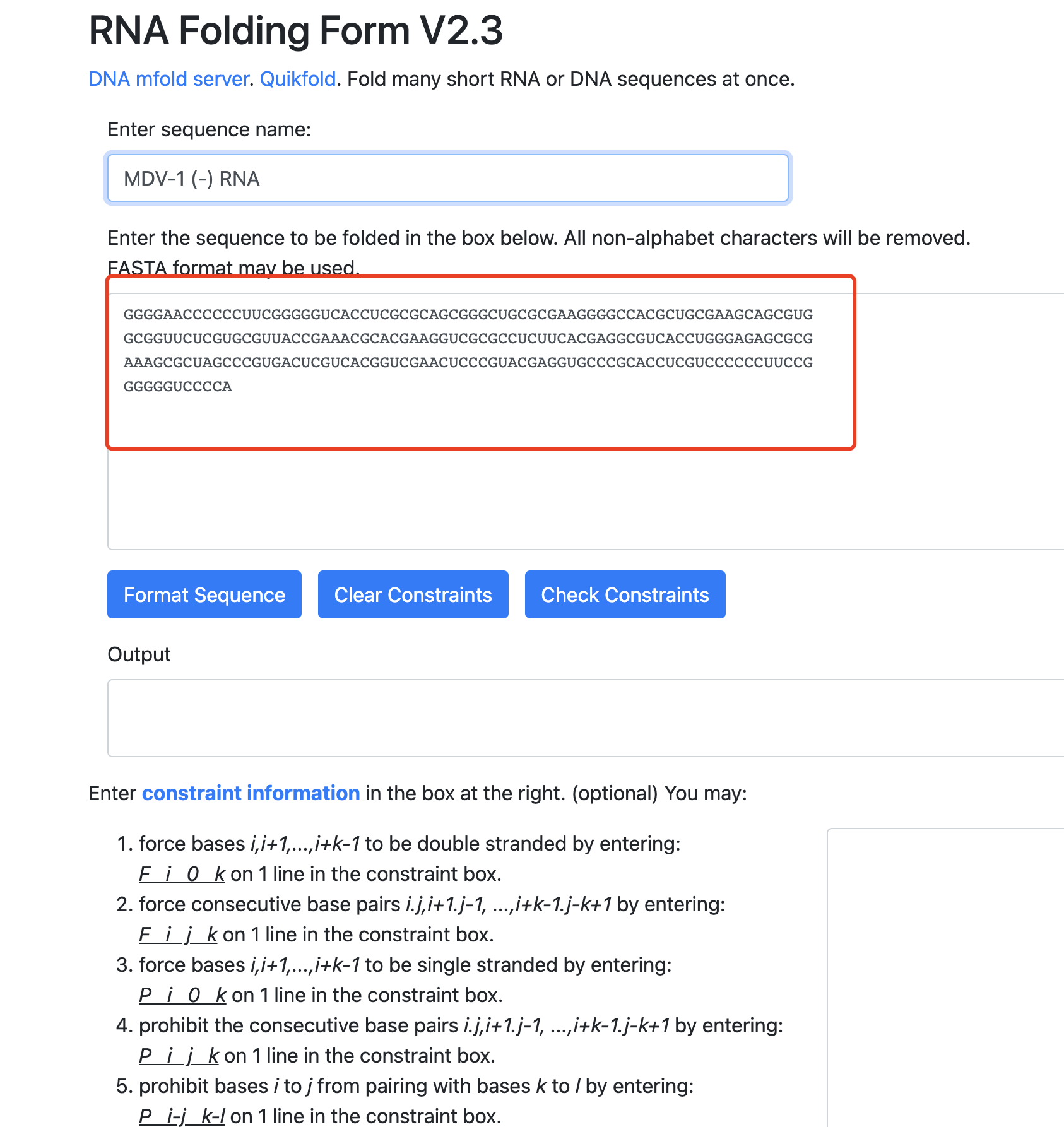
其实呢,这样的绝大部分生物信息学本科专业所教的知识点啦,但是这些呢,并不是社会实践中的生物信息学,真正的生物信息学不仅仅是能使用它这个mfold的网页工具,还需要能解决问题。比如这个网页工具是有序列输入长度限制的,如果你的序列超级长,比如最近流行的新冠病毒的某个RNA基因,就会超出限制,那么我们就得使用另外一个方法来进行mfold, 就是命令行软件使用。
很久以前我分享过1000个生物信息学软件安装,见:生物信息学常见1000个软件的安装代码!,它们是有规律的,比如对C语言的源代码软件来说,就是3部曲啦,首先,登陆你的Linux系统,然后敲入下面的命令,全部的代码如下:
mkdir -p ~/biosoft/mfold && cd ~/biosoft/mfold
wget http://www.unafold.org/download/mfold-3.6.tar.gz
tar zxvf mfold-3.6.tar.gz
cd mfold-3.6
./configure prefix=$HOME/biosoft/bin/
make
make install
# 前提是你的电脑存在 $HOME/biosoft/bin/ 文件夹,并且该 $HOME/biosoft/bin/ 文件夹是在环境变量
# 如果不在环境变量,就需要下面的2行代码哦!
PATH=$PATH:$HOME/biosoft/bin
export PATH
这样的话,你的Linux系统里面就有了mfold这个命令,如果你认真看它,其实就会发现它是一个shell脚本:
#!/bin/bash
# This shell script folds an RNA or DNA sequence and creates output
# files.
export _POSIX2_Version=0
export Package_URL="http://mfold.rna.albany.edu"
export DATDIR=`mfold_datdir|sed -e 's@/$@@'`
export Package=mfold
export Version=3.6
# Abort subroutine
abort() {
rm -f mfold.log fort.*
if [ $# -gt 0 ] ; then
echo -e "$1"
fi
echo "Job Aborted"
exit 1
}
虽然如此简单,但是没有这个基础的小伙伴仍然是会花式报错哦:

既然我们在其网页工具都是输入序列即可,软件使用理论上也是如此,前面的网页工具使用的时候,我就是输入了软件安装包里面自带的测试序列:
>MDV-1 (-) RNA
GGGGAACCCCCCUUCGGGGGUCACCUCGCGCAGCGGGCUGCGCGAAGGGGCCACGCUGCGAAGCAGCGUG
GCGGUUCUCGUGCGUUACCGAAACGCACGAAGGUCGCGCCUCUUCACGAGGCGUCACCUGGGAGAGCGCG
AAAGCGCUAGCCCGUGACUCGUCACGGUCGAACUCCCGUACGAGGUGCCCGCACCUCGUCCCCCCUUCCG
GGGGGUCCCCA
如果前面的对mfold软件的C语言的源代码安装三部曲正常的话,该软件就可以调用,先看看帮助文档,如下:
$mfold
Usage is
mfold SEQ='file_name' with optional parameters:
[ AUX='auxfile_name' ] [ RUN_TYPE=text (default) or html ]
[ NA=RNA (default) or DNA ] [ LC=sequence type (default = linear) ]
[ T=temperature (default = 37 deg C) ] [ P=percent (default = 5) ]
[ NA_CONC=Na+ molar concentration (default = 1.0) ]
[ MG_CONC=Mg++ molar concentration (default = 0.0) ]
[ W=window parameter (default - set by sequence length) ]
[ MAXBP=max base pair distance (default - no limit) ]
[ MAX=maximum number of foldings to be computed (default 100) ]
[ MAX_LP=maximum bulge/interior loop size (default 30) ]
[ MAX_AS=maximum asymmetry of a bulge/interior loop (default 30) ]
[ ANN=structure annotation type: none (default), p-num or ss-count ]
[ MODE=structure display mode: auto (default), bases or lines ]
[ LAB_FR=base numbering frequency ] [ ROT_ANG=structure rotation angle ]
[ START=5' base # (default = 1)] [ STOP=3' base # (default = end) ]
[ REUSE=NO/YES (default=NO) reuse existing .sav file ]
对于软件安装包里面自带的测试序列,我们可以这样运行:
$mfold SEQ='test.txt'
# 不知道为什么我的报错了,如下:
mfold version 3.6
REUSE= NO
test.txt.pnt created.
Sequence length is 221
Folding at 37 degrees using version 3.0 dat files.
Save file created using nafold.
Save file is empty. No foldings.
Job Aborted
好奇怪啊,我也不知道为什么我的报错了。因为这个软件并不是我的刚需,我也懒得去解决这个bug啦,如果有这方面经验的小伙伴欢迎留言交流哈!
如果是正确运行,大概会出现如下结果:
542K Mar 21 2009 mdv1.37.ct
5.1K Mar 21 2009 mdv1.37.ext
13K Mar 21 2009 mdv1.37.plot
39 Mar 21 2009 mdv1.dG
61K Oct 24 2009 mdv1.jpg
239 Nov 19 2009 mdv1-local.seq
33 Nov 19 2009 mdv1.log
51K Oct 24 2009 mdv1.pdf
14K Mar 21 2009 mdv1.plot
12K Oct 24 2009 mdv1.png
21K Oct 24 2009 mdv1.ps
109 Mar 21 2009 mdv1.run
4.0K Jan 11 16:12 mfold
相当于你在网页工具里面提交序列,选择参数,然后运行拿到的交互式结果。
比如:https://pypi.org/project/seqfold/, seqfold is an implementation of the Zuker, 1981 dynamic programming algorithm, the basis for UNAFold/mfold, with energy functions from SantaLucia, 2004 (DNA) and Turner, 2009 (RNA).
类似的,也会有R语言,java等等开发的mfold。
更高级啦,这里略。如果你感兴趣,可以考虑加入我们的《生信小成之conda交流群》。
我们邀请到了,简书conda教程单篇阅读量破40万的人气作者卖萌哥为咱们《生信技能树》和《生信菜鸟团》粉丝在钉钉群直播授课。直播是免费的哈,赶快下载钉钉软件加入吧,“Linux公益课(2021) 生物信息学”群的钉钉群号:33840083,下周六(2021-01-16)晚上八点开课哈。
同时我们提供一个微信交流群(钉钉软件我们并不是随时在线,不方便交流,钉钉仅仅是直播授课时候开启聊天),还是老规矩,18 元进群,一个简单的门槛,隔绝那些营销号!,仅此而已,考虑清楚哦! 进群方式详见公众号推文:很多事情不一定有答案(但是可以有交流渠道)
到底是各式各样的网页工具操作是生物信息学呢,还是基于Linux的NGS操作才是生物信息学呢?
太多人有这样的疑问,为什么自己进行ID转换的时候,成功率很低,今天就为你解惑。
当我们遇到了这样一个表达矩阵(其实就是从tcga数据库下载,通过ucsc的xena浏览器啦): Continue reading
前面我们的明码标价之普通转录组上游分析,受到了各大热心粉丝的吐槽,觉得太简单了我们居然还好意思收费。 Continue reading
最近在朋友圈刷到了【一刻talks先见未来大会2020】的演讲视频,其中一个讲者很有意思、就是海尔集团海创汇合伙人,首席生态官——檀林,他的演讲:数字游民时代来临,你做好准备了吗? 详细演讲视频介绍见:檀林:数字游民时代来临,你做好准备了吗? Continue reading
最近看到一个文章,标题是:《Comprehensive circular RNA profiling reveals the regulatory role of the circRNA-000911/miR-449a pathway in breast carcinogenesis》,发表于February 5, 2018 https://doi.org/10.3892/ijo.2018.4265 实验设计超级简单: Continue reading
去年我在《生信技能树》给学员分享过一个案例:GSE16561 复现,小众的illumina bead芯片,链接在:http://www.bio-info-trainee.com/7483.html ,当时大费周折才拿到跟其数据集原始文献类似的结果,已经是很满意了。最近把这个数据集作为任务安排给最新学徒们,他们反馈给我的结果让我丈二和尚摸不着头脑,居然是百分百还原文献结果,如下所示的差异基因列表: Continue reading
通常我们拿到一个全新服务器,要给大家使用,需要配置最新版R环境,代码如下:
有一些学生的Linux功底实在是太差了,所以我不得不重启六年前的《生信工程师》面试题给他们练习,有一个题目就是探索gtf。有意思的是学生们给我反馈了有几个基因居然既是lncRNA又是protein_coding。
大家应该是注意到了我们最近两周在《生信菜鸟团》紧锣密鼓的福利轰炸:
Continue reading
看到于2020年11月发表在杂志《nature cancer》的文章:《Mutations in BRCA1 and BRCA2 differentially affect the tumor microenvironment and response to checkpoint blockade immunotherapy》里面有全基因组测序数据,文献链接是:https://www.nature.com/articles/s43018-020-00139-8 Continue reading
最近共享了一些服务器,主要是唐医生的功劳,已经成功运行3台:
Continue reading
前面我们通过RcisTarget包的 cisTarget()函数,一句代码就完成了我们的hypoxiaGeneSet.txt文本文件的171个基因的转录因子注释。见:基因集的转录因子富集分析
好久没有写TCGA数据库教程了,因为TCGA计划早在2017年就陆陆续续停止了,我那个时候写了几百个教程并且录制了视频。