RNA-seq标准分析,我们已经讲解的太多了,表达矩阵到差异分析等下游生物学注释都没有啥新颖之处,融合基因和可变剪切算是出彩的地方,如果加上GATK找变异流程就更棒了,反正都使用了star软件进行序列比对拿到bam文件了。 Continue reading
一
18
RNA-seq标准分析,我们已经讲解的太多了,表达矩阵到差异分析等下游生物学注释都没有啥新颖之处,融合基因和可变剪切算是出彩的地方,如果加上GATK找变异流程就更棒了,反正都使用了star软件进行序列比对拿到bam文件了。 Continue reading
就是STAR-fusion啦,它可以直接基于STAR比对好的bam文件来做分析,而大多数其它融合基因查找工具,需要从fastq文件开始,不太方便。之前我在生信技能树公众号介绍过它,那个时候发表该工具的文章是:STAR-Fusion: Fast and Accurate Fusion Transcript Detection from RNA-Seq 在biorxiv预印本: Continue reading
最近一直在推送转录本差异相关的教程,见:每月一生信流程之rnaseqDTU(差异转录本) 扩充了大家对RNA-seq数据的理解,而且也指出来了,严格意义上的转录本定量其实是不容易的,对于二代测序来说:转录本定量本来就不是一件容易的事情 看留言,大家都深有同感! Continue reading
gtf文件大家都了解,基因或者外显子的坐标相对独立,但是转录本很不一样,同一个基因的不同转录本共用外显子,这样的话它们的坐标其实很多都是overlap的,这样,我们的二代测序的100bp或者150bp的reads就无法判定它到底属于哪一个转录本!(这个时候全长转录组测序(iso-seq)可能是更好的选择) Continue reading
最近走我整理和搭建好的:最新版针对RNA-seq数据的GATK找变异流程, 如果样本样品是正常运行,会输出: Continue reading
这是一个我也不知道该招聘什么样的人才的招聘通知!
生信技能树的粉丝都知道,**2019是我们的巡讲和宣讲元年**,是我们用实际行动来身体力行的推进生物信息学的发展,这一年我们从繁华的北上广深杭,一路狂奔到了祖国西南部的成都、重庆及西部的西安,从华北地区的天津、呼和浩特,再到华中地区的武汉、长沙、郑州,都有我们的身影! Continue reading
最近在移植一些科学界已经发表的网页小工具到我们生信技能树的服务器,发现很多工具都是依赖于rJava这个R包,在Ubuntu安装其实还是有一定的难度,所以分享一下! Continue reading
首先构建10x对象,这里就不赘述了,我在单细胞天地的2个教程:
因为计算某些基因含量这个需求实在是太常见了,所以特意设置了一个函数:PercentageFeatureSet
sce <- CreateSeuratObject(Read10X('../scRNA/filtered_feature_bc_matrix/'), "sce")
head(sce@meta.data)
GetAssayData(sce,'counts')
sce[["percent.mt"]] <- PercentageFeatureSet(sce, pattern = "^MT-")
VlnPlot(sce, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
这样就可以可视化我们计算好的线粒体基因含量,下图可以看出需要最起码的过滤。
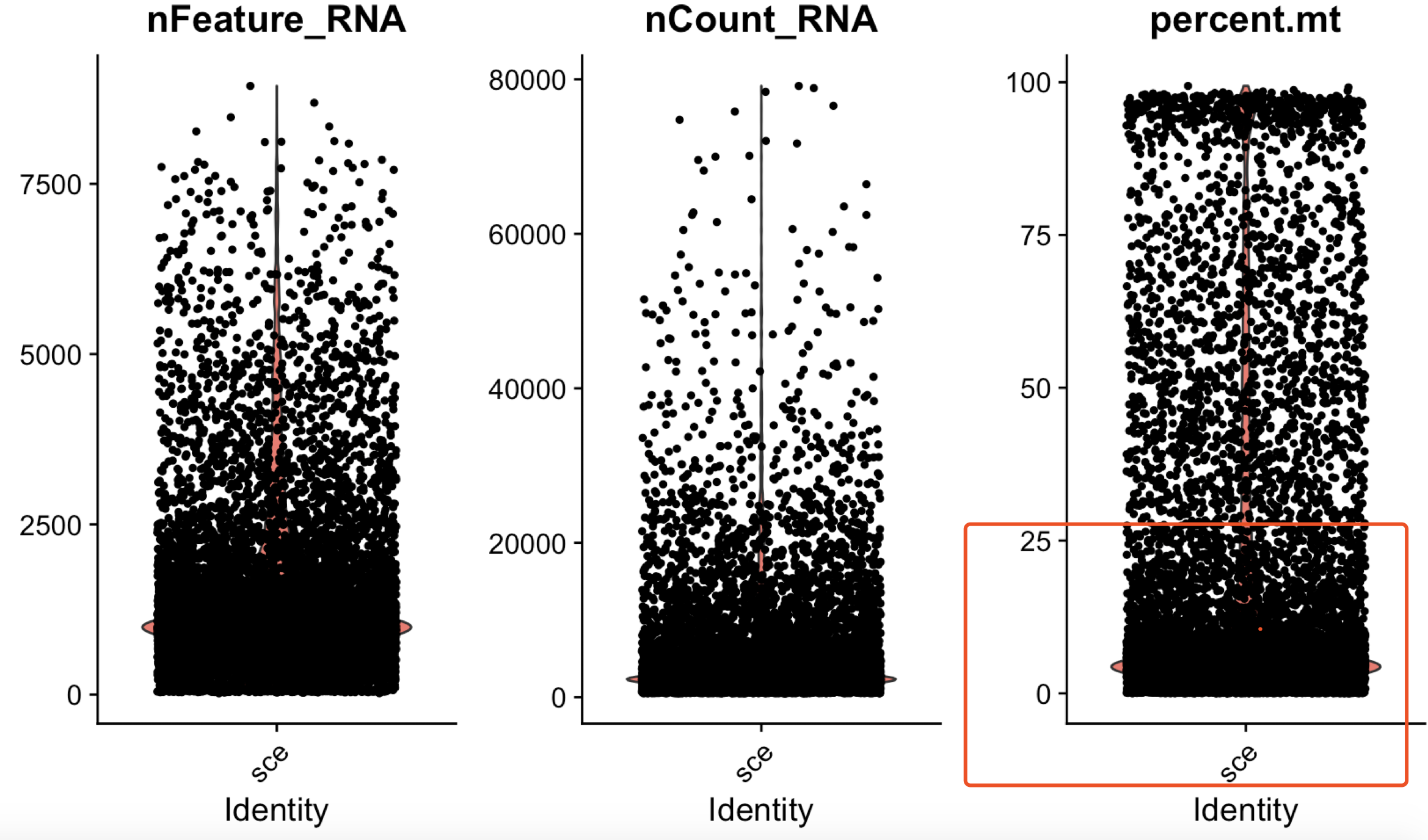
一般来说,这个过滤起码得是线粒体基因含量占比25%以下的细胞才保留,当然也得考虑到生物学课题啦。
上面的方法是修改 sce[[“percent.mt”]] ,下面我们演示 AddMetaData 函数,同样是可以增加线粒体基因含量信息到我们的seurat对象。
mt.genes <- rownames(sce)[grep("^MT-",rownames(sce))]
C<-GetAssayData(object = sce, slot = "counts")
percent.mito <- Matrix::colSums(C[mt.genes,])/Matrix::colSums(C)*100
sce <- AddMetaData(sce, percent.mito, col.name = "percent.mito")
sce[["percent.mito"]]
rb.genes <- rownames(sce)[grep("^RP[SL]",rownames(sce))]
percent.ribo <- Matrix::colSums(C[rb.genes,])/Matrix::colSums(C)*100
sce <- AddMetaData(sce, percent.ribo, col.name = "percent.ribo")
如下所示,可以看到部分细胞的核糖体基因含量也过高,至于过滤的指标,大家需要看文章啦!
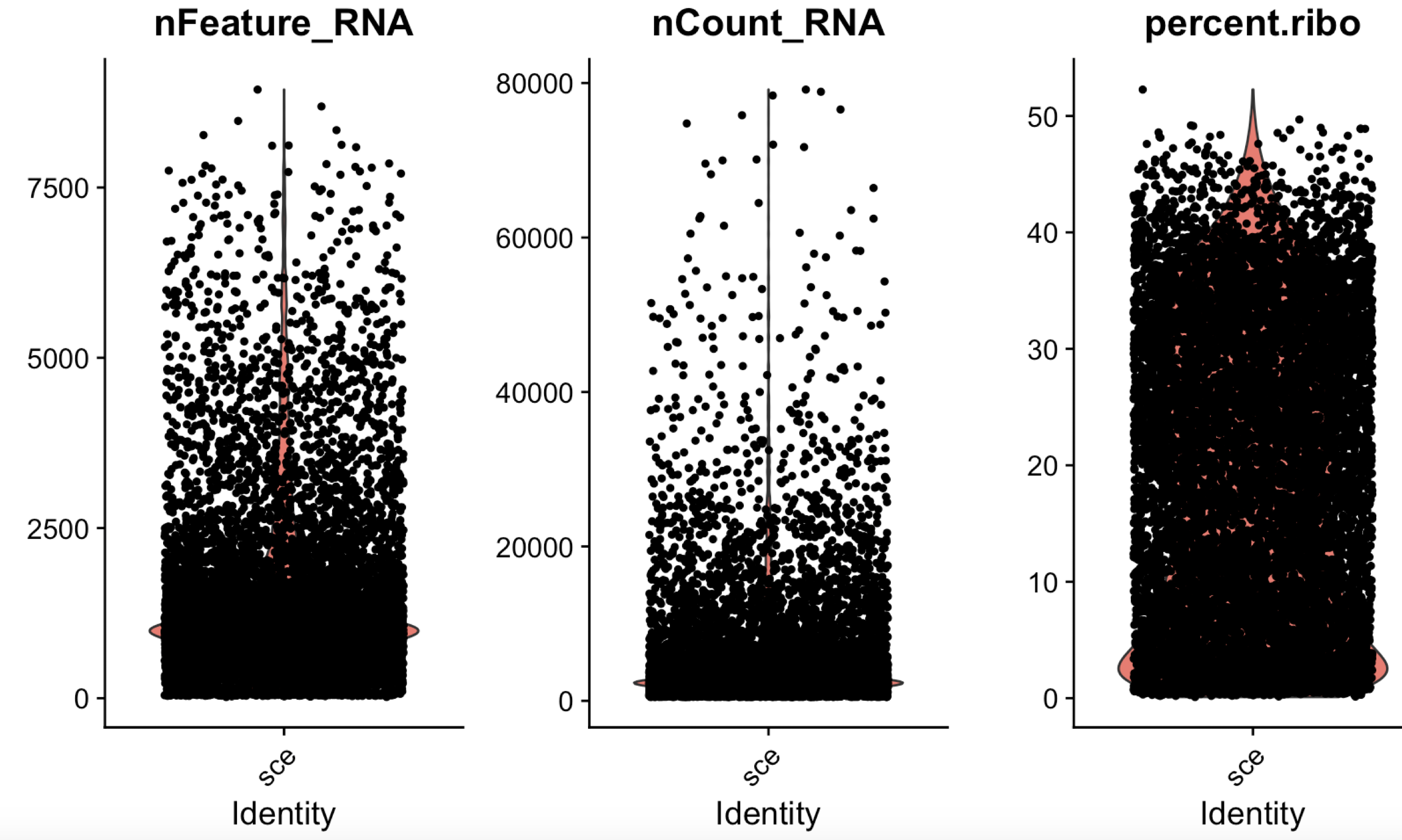
也可以是免疫球蛋白相关基因含量等等,取决于大家的生物学课题啦。
我在《生信分析人员如何系统入门Linux(2019更新版)》把Linux的学习过程分成6个阶段 ,提到过每个阶段都需要至少一天以上的学习: Continue reading
朋友圈医务工作者不少,经常看到各个疾病方向的义诊通知,各大城市均有,很佩服大家,而且我表示实名羡慕。虽然我不是学医的,但是也可以从另外一个层面帮助一下大家,我也来一个义诊,那就是我们生信技能树最擅长生物信息学方面的“义诊”啦! Continue reading
昨天我们重点强调了star这个比对软件开发团队,附带的star-fusion:最好用的融合基因查找工具终于正式发表了 因为我自己是时隔两年后再次使用它,所以很多数据库和软件代码都没有更新,中间一个小报错就浪费了四五个小时,所以分享一下这个体验! Continue reading
最近下载一篇文章的数据,发现3个数据,就有3种结果: Continue reading
因为在中国大陆安装R包,通常是切换镜像的,我会首先推荐清华的镜像给学生们,切换镜像的代码如下: Continue reading
看到九月份学徒在群里提问,写爬虫批量循环抓取NBCI数据库的基因信息,但是经常掉线,还有可能被封,求助!
我简单指点了他去找基因数据库文件即可,随便邀请他总结投稿如下: Continue reading
最近在调试gatk流程的时候,发现一个很有趣的问题,我使用gvcf模式的Variant Calling的代码如下: Continue reading
今天的GEO数据挖掘课程,有一个学员问到在向量的任何位置插入任何元素有没有什么简介的方法,因为她做的很麻烦,如下:
有一个向量,是100个元素,要在第34位加上一个数是56 Continue reading
2019上半年我抽空在各个周六日走了十几个城市,详情见:走过了12个城市,接下来去哪里遇见你 中间几个月博士中期考核实在是太忙就暂停了全国分享的脚步。现在重启,发现继续命名为五周年可能不合适了,因为已经进入了第6年了,这次我们一次性公布3个城市的宣讲会: Continue reading
最近接到TCGA分析需求,想看看指定基因在指定癌症是否具有临床意义(也就是生存分析是否有统计学显著效果咯!)其实很早以前我在生信技能树就号召粉丝讨论过这个问题:集思广益-生存分析可以随心所欲根据表达量分组吗 这里我做实力演绎一下。 Continue reading
我们多次在生信技能树公众号介绍过star-fusion这个目前最好的针对RNA-seq测序数据找融合基因的软件:最好用的融合基因查找工具终于正式发表了 ,还有一个踩过的坑需要注意:[一个好像没有做任何改变的参数] Continue reading
通常我们讲解GEO数据挖掘,指的是表达芯片数据处理,其中一个难点就是芯片设计的探针跟我们感兴趣的基因的对应关系,之所以说它是难点,就是背景知识太多,初学者无从了解。 Continue reading