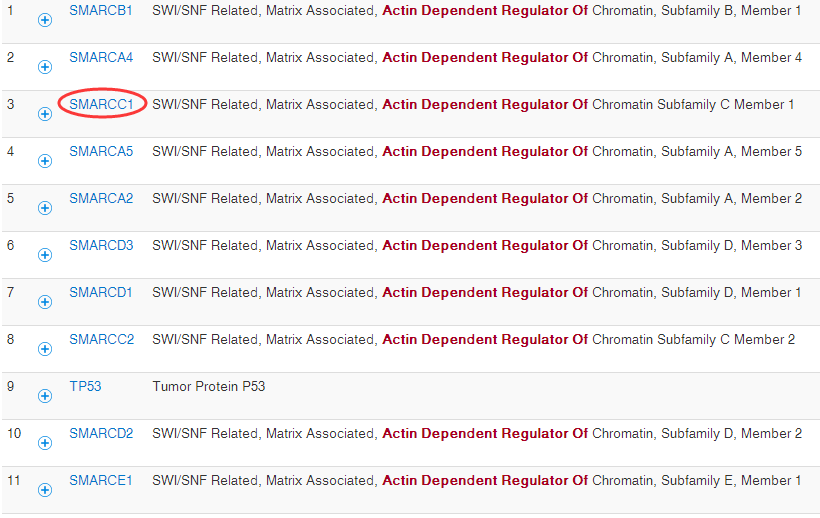
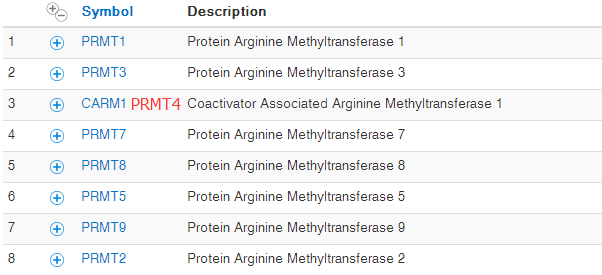
可变剪切是指mRNA前体以多种方式将exon连接在一起的过程。 由于可变剪切使一个基因产生多个mRNA转录本,不同mRNA可能翻译成不同蛋白。
十一
09
基因融合(Gene fusion)是指将两个或多个基因的编码区首尾相连,置于同一套调控序列(包括启动子、增强子和终止子等)的控制之下,构成嵌合基因。基因融合通常是由于染色体重排所造成的。异常基因融合事件可以引起恶性血液疾病以及肿瘤的发生,所以通过分析基因融合现象将有助于探讨发病机制、biomaker的筛选等,临床意义重大。 Continue reading
其实这个植物是拟南芥,所以跟人类研究的数据处理大同小异。
转录组测序的研究对象为特定细胞在某一功能状态下所能转录出来的所有 RNA 的总和,包括 mRNA 和非编码 RNA 。通过转录组测序,能够全面获得物种特定组织或器官的转录本信息,从而进行转录本结构研究、变异研究、基因表达水平研究以及全新转录本发现等研究。 Continue reading
本次实践是根据Jimmy 和洲更两位大神的笔记,还有Y叔的Chipseeker包,第一次跑Chip-seq流程,主要关注了流程能否跑通,怎么解决自己实践中的Bug。 Continue reading
https://canvasxpress.org/html/index.html 这个包里面有好多比较漂亮的模版推荐给大家!
下面的图片点击可以放大,从上面的链接还可以进入查看R代码,数据,完整的重复出来该可视化图片 Continue reading
首先上目录: Continue reading
近年来,单细胞生物学研究一直是热点领域,而尤为大家所关注的就是单细胞转录组测序了(single-cell RNA sequencing (scRNA-seq)). Continue reading
WGCNA其译为加权基因共表达网络分析。该分析方法旨在寻找协同表达的基因模块(module),并探索基因网络与关注的表型之间的关联关系,以及网络中的核心基因。
适用于复杂的数据模式,推荐5组(或者15个样品)以上的数据。一般可应用的研究方向有:不同器官或组织类型发育调控、同一组织不同发育调控、非生物胁迫不同时间点应答、病原菌侵染后不同时间点应答。 Continue reading
github极简指南
入生信的坑已经3年多了,但是开始github的旅程才一年多,起初主要是为了建立bioconductor中文社区而学习的,现在也在自己的github上面分享了不少代码,有一些心得体会,欢迎大家前往github star我的项目
今天又有小伙伴微信问我perl模块安装的问题,因为ENSEMBL发布的大多数数据库以及软件都是基于perl的,尤其是分量很重的VEP,所以即使你再如何如何的讨厌perl,也不得不与之打交道。
这种细节问题问我,我当然无法直接给出答案咯。毕竟,我的知识积累都不是靠死记硬背的。所以需要取回过头查看一下我的博客,才意识到,我竟然已经写了7篇教程,关于perl的模块。目录如下:
首先需要自己确定已经安装了哪些模块,都安装在哪里?还有新的模块需要安装到哪里?
然后再学习如何安装新的模块。
DIPG和 adult glioblastomas (GBMs) 很大区别,不应该用同样的治疗方式。
测序策略是:
We integrated deep sequencing analysis of 36 tumor-normal pairs (20 whole-genome sequencing (Illumina HiSeq 2000) and 16 whole-exome sequencing (Applied Biosystems SOLiD 5500xl)) with comprehensive methylation (28 DIPGs; Illumina Infinium450k methylation array), copy number (45 DIPGs; Affymetrix SNP6.0) and expression (35 DIPGs; Illumina HT-12 v4) data (Supplementary Table 1). Continue reading
很久很久以前我就写过一个服务器系列教程:http://www.bio-info-trainee.com/555.html
在那里,我留下了一个疑问,因为后来没有机会再继续捣鼓服务器,所以一直悬而未决,问题描述如下: Continue reading
我知道会有续集,但也没想到续集来得这么快!今天收到了一个生信技能树公众账号铁杆粉丝(我们之间有过9次邮件交流)的求助信,下面我们首先一起帮他解决一下碰到的问题。随后和大家分享一下可以提高搜索效率和准确率的Google搜索技巧。
今天是大年初二,这篇文章我只想传达一点:
没有什么菜鸟级别的生物信息学数据处理是不能通过Google得到解决方案的,如果有,请换个关键词继续Google!
首先用两分钟的时间简单介绍一下R语言:
因为这个语言是肉丝儿(Ross Ihaka)和萝卜特(Robert Gentleman)两个人1992年在S语言的基础上发明出来的开源语言,所以叫做R语言。这两个人是统计学教授出身,所以R语言在统计学方面有着纯正的血统!如果你平时的工作和统计相关,你好意思不会点R语言么?
垃圾数据对初学者的伤害真的很可怕!
最近在带一些朋友入门,想起了当年自己入门的各种凄惨惨戚戚!
碱基质量值很差,GC不平衡,还有接头,PCR重复也很多,kmer值也很诡异,时间都耗在QC上面了,结果几个月下来,你一个流程都没搞明白,各种查资料,还是在原地打转。 Continue reading
我们在第19讲留下了一个悬念:为什么我算的染色体覆盖度与公司给我的报告不符合!!
公司算出每条染色体的覆盖度都接近于100%了,而我是结果是: Continue reading
来自于我们生信技能树论坛的超级版主bioinfo.dong的好文一篇,比较符合我博客的思想,就友情转发一下:
原文链接见:http://www.biotrainee.com/thread-379-1-1.html
刚接触生信的同学大都有个困惑,知道生物信息可能需要编程,可是选择什么语言呢?有人会说perl啊,Python啊,R啊,java啊,等等等等。目的不一样,选择也不一样,你可以说语言都没有区别,达到目的就行,当然没问题。可是我们也要知道每种语言都有其独特优势,你可以用perl倒腾出矩阵运算,也可以画出想要的图,可是没有R专业;你也可以用R的正则表达式处理文本,可是perl或者Python做正则会更方便一些。这不是比较帖,只是从一个Python体验者的角度来说一下为什么选择Python。我目前的编程组合是Python+R+Shell Scripting。
这篇文章比较适合编程初学者,常年用perl的老司机们可以随便看一下,虽说perl和Python很像,有了一门的基础,学另一门就容易多了,可是真让一个用了几年perl的人彻底换Python还是比较困难的,主要还是习惯问题。最初做生信的人大都以perl作为常用脚本语言。我也是从perl开始的,当年为了申请出国读Bioinformatics,认真把小骆驼书看了一遍。来美国之后的第一个导师刚好是教perl的,我又跟着学了一次,看完导师推荐的《Unix and Perl to the Rescue》,算是巩固加第二次入门。之后一年基本都是用perl来处理数据。一个偶然的机会,同学说一起学学Python吧,听说很好用,于是就在网上找了个教程把题目刷了一遍。虽说入了门,可是每次项目赶时间的时候第一个想到的还是用perl来解决,所以入门很久也没啥长进,我亲爱的同学因为perl用的太好,虽然知道Python很好用,可始终没法狠心转过来,而我因为本身perl学得也只是半斤八两,纠结了一段时间也就彻底放弃perl了。
先说用了很长时间perl再用Python觉得不习惯的点。
(1)首先是动物园的书,《learning perl》真是入门的典范。再看《Learning Python》,几千页,那么厚,我到现在也没法认真看下去。
(2)另外perl语句比较简洁,几个符号就可以讲清楚的,Python可能需要几行,比如按行读取,perl只要while(<>)就可以,而最初学Python的时候,光这个问题就困扰了很久。再比如perl正则匹配的$1, Python是match.group(1)。perl的简洁伴随的缺点是可读性较差,自己的代码写完了都不想再看,更不要说别人写的。
(3)perl的正则表达式是真的非常厉害,我已经不记得是怎么厉害的了,就只记得Python的re module刚开始接触不太好用,不过现在已经感觉不出区别了。
(4)通常一个Python脚本需要很多modules,不熟悉之前会觉得很痛苦,perl就比较少用到,我总共也没用几次,一方面说明我的perl确实学得不好,另一方面可能也真是不太好用,看到就觉得麻烦。但Python的modules一旦熟悉了会大大提高工作效率。重点说一下Python的优点。Python作为编程语言真正的优势比如面向对象编程(OOP),可移植/扩展/嵌入,强大的爬虫功能,APP开发,web开发等都不在讨论范围之内,只从最实用的角度做一下说明:
(1)简单,适合作为入门语言。很多时候觉得读Python的代码像是在读简单的英文,或者觉得pseudocode稍微一改就可以在Python里run了。Python还规范了很好的写作格式,该缩进的必须缩进,这样更增强了可读性。同时提高了代码重复利用的可能(很多时候perl代码写完就不想读了,三个月不用再回来已经看不太懂了,Python的就可以留着慢慢用。。。)
(2)Python社区活跃。有问题可以很容易搜索到解决方案。我perl的老师现在也转教Python了,问他为什么,他说perl的community不活跃,用Python是一种趋势
(3)作为开源语言,Python有很多非常好用的包,可以最大程度让我们避免把时间浪费在重复造轮子上。刚接触Python的时候我就觉得这简直是perl和R的整合,之前提过Python的scipy,numpy,pandas,matlibplot等等packages使其同样拥有了很强大的统计画图功能,我曾一度弃用R,用Python做所有的数据处理,数据分析和画图。不过现在又将这些工作交回了R,实验室本身是做统计的,用R显得入流一点:-)
(4)Python的jupyter notebook!!!这个是要强力推荐的!!!以前叫ipython notebook。用过R的都知道R Studio。jupyter notebook就是Python的Studio。以前写perl或者Python是不是这样的流程:写好了,存成.pl或.py格式,在shell里python xxx.py或者perl xxx.pl。运行完发现不好,有bug,打开文件找找bug在哪,再运行,还不行,唉,反反复复,好累。有了jupyter notebook你就可以边写边跑边改程序。有任何不确定的地方,都可以在notebook里直接测试,有任何bug都可以在notebook里直接改。简直方便到爆。现在用Anaconda安装jupyter还附赠很多包,方便又实惠。
(5)学好Python可以转行!!!跳出生物坑,奔向美好的互联网坑。前面提到的爬虫,APP开发,web编程都是很实用的技能。许多互联网公司也会专门招Python程序员,比如Google,比如Youtube,比如Dropbox。。。
我本专业是Bioinformatics,需要上一些计算机和统计的研究生课程,还记得算法课上老师第一节课就问,java和C++都会吧,如果不会的话Python总会吧,都不会的话这门课的作业写不了。就因为觉得自己还算会一点Python,把一次学习java的好机会浪费掉了
暂时就想到这么多。说的未必对。都是自己的体会吧。希望对初学者有用~
因为最近在研究CHIP-seq测序数据处理,发现有些文章重点并不是数据处理本身,而是对生物学基础知识的掌控以及实验设计,这篇文章我重点推荐一下,我略微翻译了一些,笔记如下: