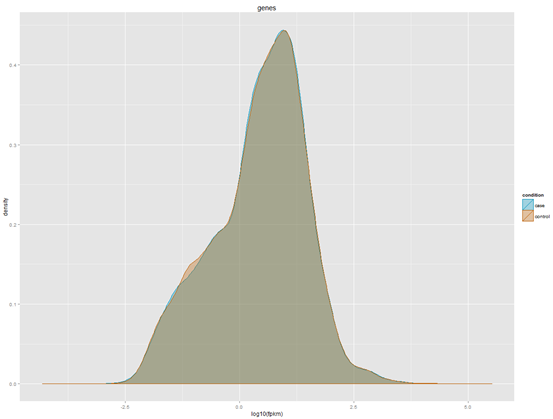
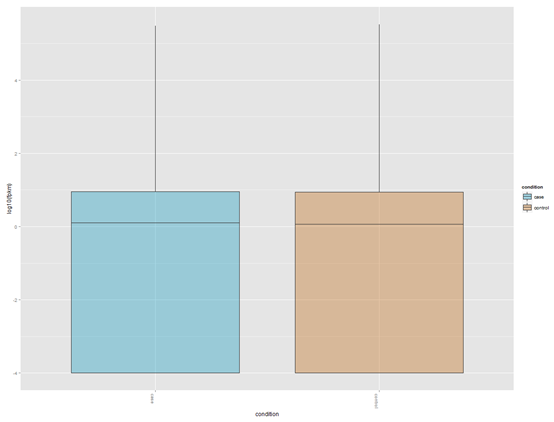
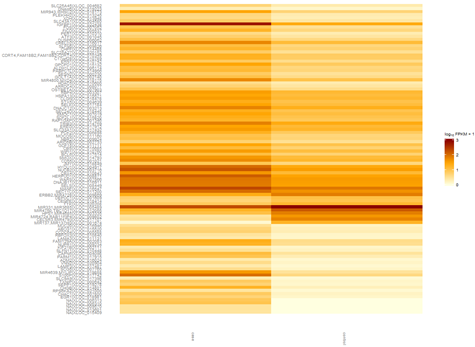
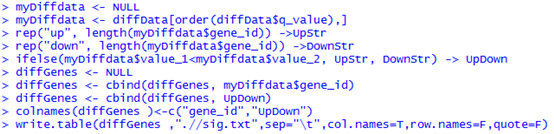
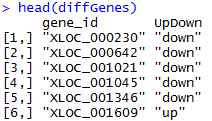
找橡胶测序数据无果
所以我只好找了他们所参考的草莓(strawberry, Fragaria vesca (2n = 2x = 14),a small genome (240 Mb),)的文章,是发表是nature genetics上面的
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3326587/
可以看到它的SRA索取号。

草莓组装结果:Over 3,200 scaffolds were assembled with an N50 of 1.3 Mb .
Over 95% (209.8 Mb) of the total sequence is represented in 272 scaffolds.
草莓基因息:Gene prediction modeling identified 34,809 genes, with most being supported by transcriptome mapping.
草莓染色体信息:Paradoxically, the small basic (x = 7) genome size of the strawberry genus, ~240 Mb,
offers substantial advantages for genomic research.
草莓来源:diploid strawberry F. vesca ssp. vesca accession Hawaii 4
(National Clonal Germplasm Repository accession # PI551572).
然后我去NCBI上面下载这三个数据
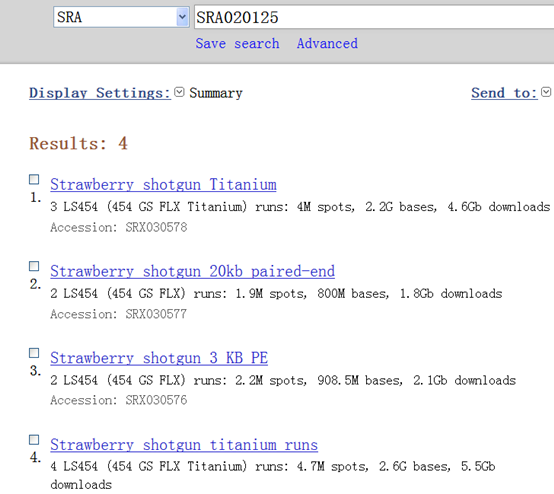
SRA020125 共有四个数据:
| http://www.ncbi.nlm.nih.gov/sra/SRX030575[accn] | Total: 4 runs, 4.7M spots, 2.6G bases, 5.5Gb |
| http://www.ncbi.nlm.nih.gov/sra/SRX030576[accn] (3 KB PE) | Total: 2 runs, 2.2M spots, 908.5M bases, 2.1Gb |
| http://www.ncbi.nlm.nih.gov/sra/SRX030577[accn] (20KB片段) | Total: 2 runs, 1.9M spots, 800M bases, 1.8Gb |
| http://www.ncbi.nlm.nih.gov/sra/SRX030578[accn] | Total: 3 runs, 4M spots, 2.2G bases, 4.6Gb |
挂在后台自动下载
![]()
好了,有了这些数据我们就要进行基因组的一系列分析啦!!!
不过我们可以先看看他们这个研究小组的成果
首先他们建造了一个关于草莓的基因组信息网站
https://strawberry.plantandfood.co.nz/
跟我之前在水科院做鲫鱼鲤鱼的差不多
直接在里面就可以下载他们做好的所有数据,也可以可视化。
它的染色体如下,非常简单,就七条染色体
http://www.rosaceae.org/species/fragaria/fragaria_vesca/genome_v1.1
我找到了它组装好的草莓基因组地址,用批处理全部下载了