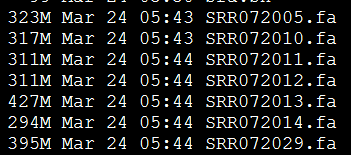
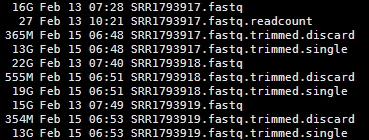
今天先 对7个单端数据做处理,是454数据,平均长度300bp左右,明天再处理3KB和20KB的配对reads。
首先跑fastqc
打开一个个看结果
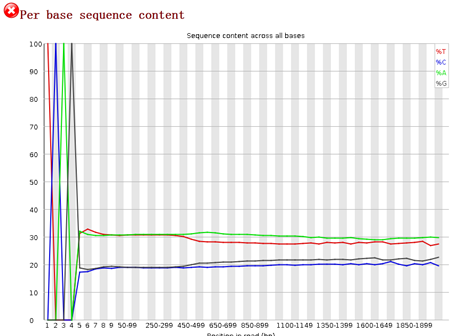
可以看到前面一些碱基的质量还是不错的, 因为这是454平台测序数据,序列片段长度差异很大,一般前四百个bp的碱基质量还是不错的,太长了的测序片段也不可靠
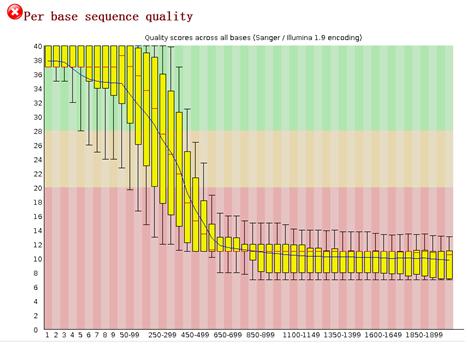
重点在下面这个图片,可以看到,前面的4个碱基是adaptor,肯定是要去除的,不是我们的测序数据。是TCAG,需要去除掉。
所以我们用了 solexaQA 这个套装软件对原始测序数据进行过滤
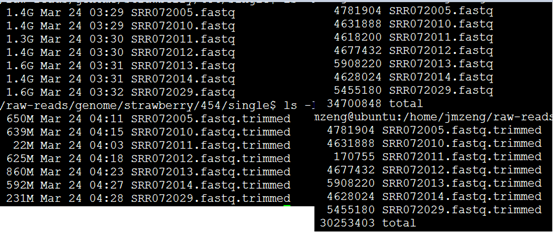
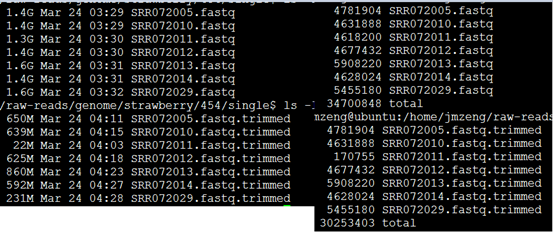
可以看到过滤的非常明显!!!甚至有个样本基本全军覆没了!然后我查看了我的批处理脚本,发现可能是perl DynamicTrim.pl -454 $id这个参数有问题
for id in *fastq
do
echo $id
perl DynamicTrim.pl -454 $id
done
for id in *trimmed
do
echo $id
perl LengthSort.pl $id
done
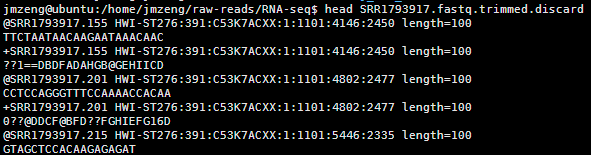
可以看到末尾的质量差的碱基都被去掉了,但是头部的TCAG还是没有去掉。
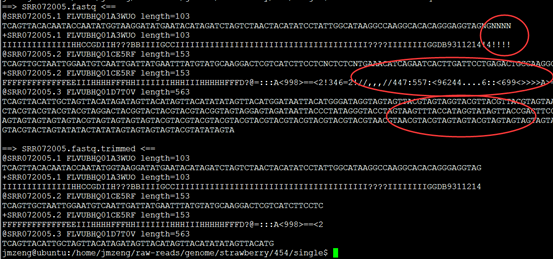
处理完毕后的数据如下: