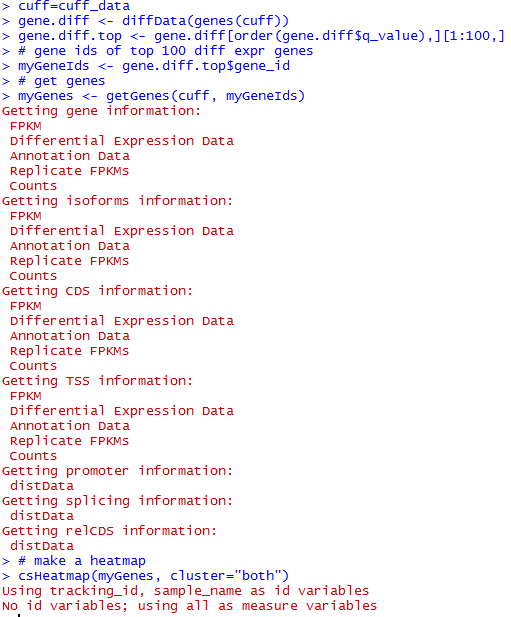
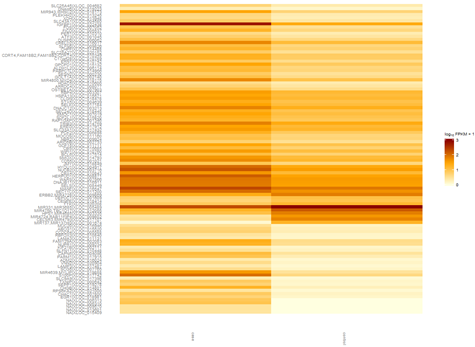
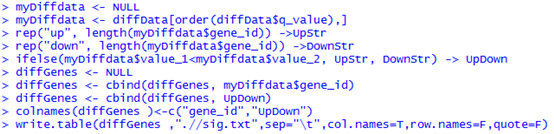
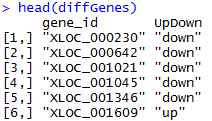
想了想,既然是菜鸟教程,那就索性再介绍点更基础的东西,基本上只要是大学毕业的都能看懂,不需要懂计算机了。首先讲讲linux服务器吧,因为生物信息也算是半个大数据分析,所以我们平常的办公电脑一般都是不能满足需求的,大部分实验室及公司都会自己配置好服务器给菜鸟们用,菜鸟们首先要拿到服务器的IP和高手给你的用户名和密码。
一般我们讲服务器,大多是linux系统,而我这里所讲的linux系统呢,特指ubuntu,其余的我懒得管了,大家也不要耗费无谓的时间纠结那些名词的不同!
登录到服务器有两种方法,一种是ssh,传输你的命令给服务器执行,另一种是ftp,和服务器交换文件。而ssh我们通常用putty,xshell等等。ftp呢,我们可以用winscp,xshell,所以我一直都用xshell,因为它两者都能搞定!
Xshell软件自行搜索下载,打开之后新建一个连接,然后登陆即可。
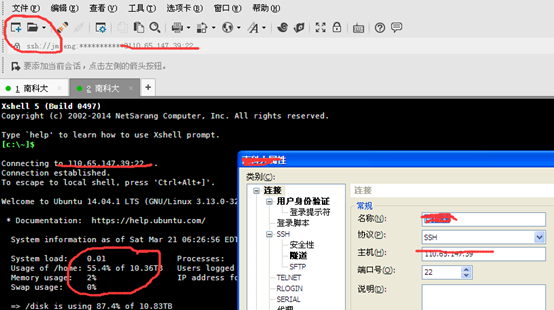
然后输入以下命令,可以查看服务器配置,包括cpu。内存,还有硬盘
cat /proc/cpuinfo |grep pro|wc -l
free -g
df -h
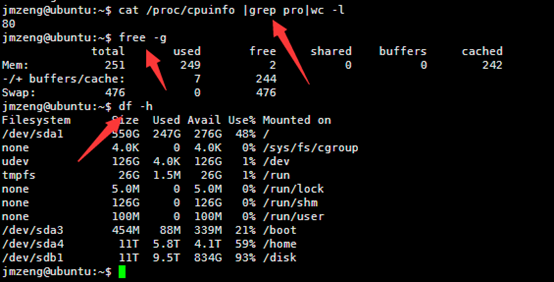
这个服务器配置好一点,有80个cpu,内存256G,硬盘有2个11T的,是比较成熟的配置。
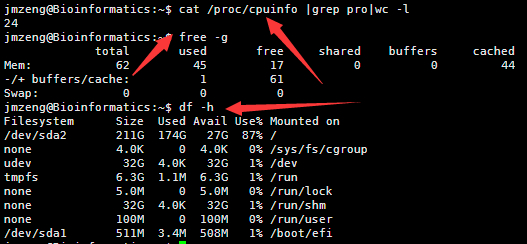
这个是一个小型服务器。也就24个核,64G的内存,但是存储量有点小呀,其实可以随便花几百块钱买个1T的硬盘挂载上去的。
然后linux的其它命令大家就得自己去搜索一个个使用,然后熟悉,记牢,然后创新啦!
我随便敲几个我常用的吧: ls cd mkdir rm cp cat head tail more less diff grep awk sed grep perl 等等!
呀,突然间发现我才介绍了ssh的方法登陆服务器并且发送命令在服务器上面运行,下面贴图如何传输文件。一般xshell的菜单里面有绿的文件夹形式的标签就是打开ftp文件传输,这种可视化的软件,大家慢慢摸索吧!