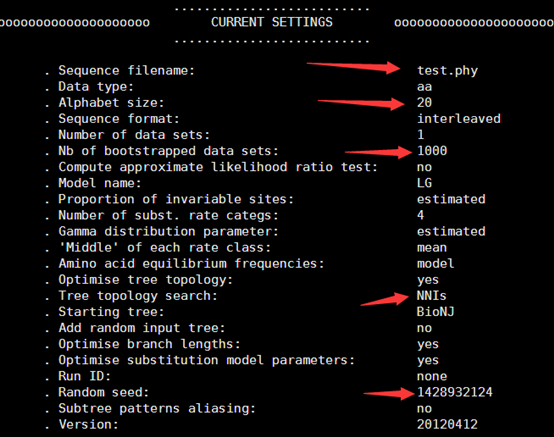
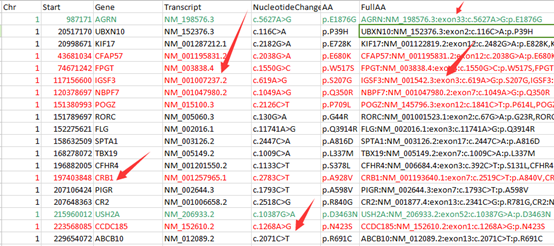
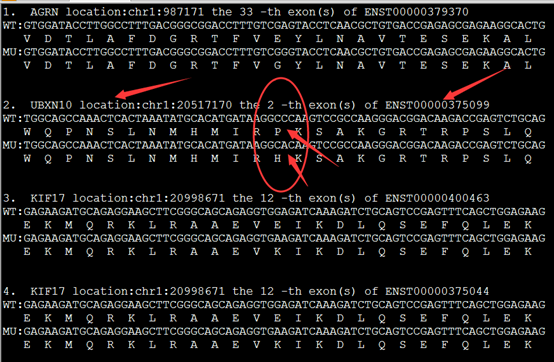
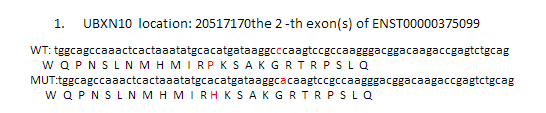因为要讲搜索,所以我选择了一个长一点的字符串来演示多种情况的搜索
perl rotation_one_by_one.pl atgtgtcgtagctcgtnncgt
程序运行的结果如下
$ATGTGTCGTAGCTCGTNNCGT 21
AGCTCGTNNCGT$ATGTGTCGT 9
ATGTGTCGTAGCTCGTNNCGT$ 0
CGT$ATGTGTCGTAGCTCGTNN 18
CGTAGCTCGTNNCGT$ATGTGT 6
CGTNNCGT$ATGTGTCGTAGCT 13
CTCGTNNCGT$ATGTGTCGTAG 11
GCTCGTNNCGT$ATGTGTCGTA 10
GT$ATGTGTCGTAGCTCGTNNC 19
GTAGCTCGTNNCGT$ATGTGTC 7
GTCGTAGCTCGTNNCGT$ATGT 4
GTGTCGTAGCTCGTNNCGT$AT 2
GTNNCGT$ATGTGTCGTAGCTC 14
NCGT$ATGTGTCGTAGCTCGTN 17
NNCGT$ATGTGTCGTAGCTCGT 16
T$ATGTGTCGTAGCTCGTNNCG 20
TAGCTCGTNNCGT$ATGTGTCG 8
TCGTAGCTCGTNNCGT$ATGTG 5
TCGTNNCGT$ATGTGTCGTAGC 12
TGTCGTAGCTCGTNNCGT$ATG 3
TGTGTCGTAGCTCGTNNCGT$A 1
TNNCGT$ATGTGTCGTAGCTCG 15
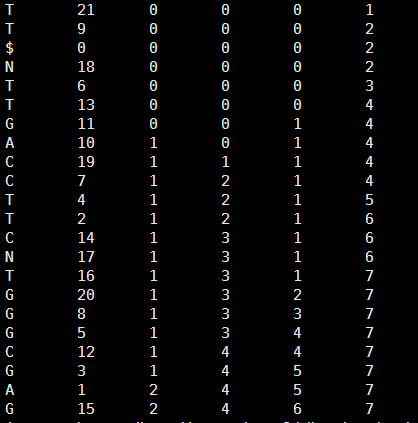
它的BWT及索引是
T 21
T 9
$ 0
N 18
T 6
T 13
G 11
A 10
C 19
C 7
T 4
T 2
C 14
N 17
T 16
G 20
G 8
G 5
C 12
G 3
A 1
G 15
然后得到它的tally文件如下
接下来用我们的perl程序在里面找字符串
第一次我测试 GTGTCG 这个字符串,程序可以很清楚的看到它的查找过程。
perl search_char.pl GTGTCG tm.tally
your last char is G
start is 7 ; and end is 13
now it is number 5 and the char is C
start is 3 ; and end is 6
now it is number 4 and the char is T
start is 17 ; and end is 19
now it is number 3 and the char is G
start is 10 ; and end is 11
now it is number 2 and the char is T
start is 19 ; and end is 20
now it is number 1 and the char is G
start is 11 ; and end is 12
It is just one perfect match !
The index is 2
第二次我测试一个多重匹配的字符串GT,在原字符串出现了五次的
perl search_char.pl GT tm.tally
your last char is T
start is 15 ; and end is 22
now it is number 1 and the char is G
start is 8 ; and end is 13
we find more than one perfect match!!!
8 13
One of the index is 11
One of the index is 10
One of the index is 19
One of the index is 7
One of the index is 4
One of the index is 2
One of the index is 14
惨了,这个是很严重的bug,不知道为什么,对于多个匹配总是会多出那么一点点的结果。
去转换矩阵里面查看,可知,前面两个结果11和10是错误的。
CTCGTNNCGT$ATGTGTCGTAG 11
GCTCGTNNCGT$ATGTGTCGTA 10
GT$ATGTGTCGTAGCTCGTNNC 19
GTAGCTCGTNNCGT$ATGTGTC 7
GTCGTAGCTCGTNNCGT$ATGT 4
GTGTCGTAGCTCGTNNCGT$AT 2
GTNNCGT$ATGTGTCGTAGCTC 14
最后我们测试未知字符串的查找。
perl search_char.pl ACATGTGT tm.tally
your last char is T
start is 15 ; and end is 22
now it is number 7 and the char is G
start is 8 ; and end is 13
now it is number 6 and the char is T
start is 19 ; and end is 21
now it is number 5 and the char is G
start is 11 ; and end is 12
now it is number 4 and the char is T
start is 20 ; and end is 21
now it is number 3 and the char is A
start is 2 ; and end is 3
now it is number 2 and the char is C
start is 3 ; and end is 3
we can just find the last 6 char ,and it is ATGTGT
原始字符串是ATGTGTCGTAGCTCGTNNCGT,所以查找的挺对的!!!
[perl]
$a=$ARGV[0];
$a=uc $a;
open FH,"<$ARGV[1]";
while(<FH>){
chomp;
@F=split;
$hash_count_atcg{$F[0]}++;
$hash{$.}=$_;
# the first line is $ and the last char and the last index !
}
$all_a=$hash_count_atcg{'A'};
$all_c=$hash_count_atcg{'C'};
$all_g=$hash_count_atcg{'G'};
$all_n=$hash_count_atcg{'N'};
$all_t=$hash_count_atcg{'T'};
#print "$all_a\t$all_c\t$all_g\t$all_t\n";
$len_a=length $a;
$end_a=$len_a-1;
#print "your query is $a\n";
#print "and the length of your query is $len_a \n";
$after=substr($a,$end_a,1);
#we fill search your query from the last char !
if ($after eq 'A') {
$start=2;
$end=$all_a+1;
}
elsif ($after eq 'C') {
$start=$all_a+1;
$end=$all_a+$all_c+1;
}
elsif ($after eq 'G') {
$start=$all_a+$all_c+1;
$end=$all_a+$all_c+$all_g+1;
}
elsif ($after eq 'T'){
$start=$all_a+$all_c+$all_g+$all_n+1;
$end=$all_a+$all_c+$all_g+$all_t+$all_n+1;
}
else {die "error !!! we just need A T C G !!!\n"}
print "your last char is $after\n ";
print "start is $start ; and end is $end \n";
foreach (reverse (1..$end_a)){
$after=substr($a,$_,1);
$before=substr($a,$_-1,1);
($start,$end)=&find_level($after,$before,$start,$end);
print "now it is number $_ and the char is $before \n ";
print "start is $start ; and end is $end \n";
if ($_ > 1 && $start == $end) {
$find_char=substr($a,$_);
$find_len=length $find_char;
print "we can just find the last $find_len char ,and it is $find_char \n";
#return "miss";
last;
}
if ($_ == 1) {
if (($end-$start)==1) {
print "It is just one perfect match ! \n";
my @F_start=split/\s+/,$hash{$end};
print "The index is $F_start[1]\n";
#return $F_start[1];
last;
}
else {
print "we find more than one perfect match!!!\n";
print "$start\t$end\n";
foreach (($start-1)..$end) {
my @F_start=split/\s+/,$hash{$_};
print "One of the index is $F_start[1]\n";
}
#return "multiple";
last;
}
}
}
sub find_level{
my($after,$before,$start,$end)=@_;
my @F_start=split/\s+/,$hash{$start};
my @F_end=split/\s+/,$hash{$end};
if ($before eq 'A') {
return ($F_start[2]+1,$F_end[2]+1);
}
elsif ($before eq 'C') {
return ($all_a+$F_start[3]+1,$all_a+$F_end[3]+1);
}
elsif ($before eq 'G') {
return ($all_a+$all_c+1+$F_start[4],$all_a+$all_c+1+$F_end[4]);
}
elsif ($before eq 'T') {
return ($all_a+$all_c+$all_g+$all_n+1+$F_start[5],$all_a+$all_c+$all_g+1+$all_n+$F_end[5]);
}
else {die "error !!! we just need A T C G !!!\n"}
}
[/perl]
原始字符串是atgtgtcgtagctcgtnncgt