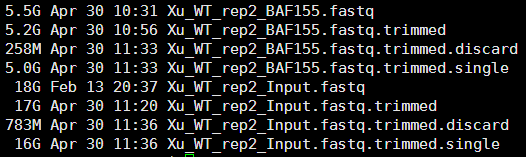
CD分子吧,它是Clusters of Differentiation的简写,是指一组分化抗原的家族,目前该家族已经有CD1——CD350甚至更多的成员.他们分布于T细胞等免疫细胞表面,参与免疫细胞各种表达,其中有整合素、受体、配体等蛋白分子,在免疫应答反应中参与识别、粘附和信号转导等功能.
我这里简单讲讲如何整理它们的基因信息,首先从NCBI里面下载的人的gene_info文件,然后通过脚本来查找CD分子信息。
perl -alne '{if (/\tCD\d+/ or /CD\d+\|/ ) {print}}' human_gene_info >CD.info
cut -f 2-5 CD.info >CD.table
再根据CD分子的排序把我们的信息重新排序
perl -alne '{/CD(\d+\w)/;$hash{$1}=$_}END{print $hash{$_} foreach sort {$a <=> $b}keys %hash}' CD.table >CD.table.sort
然后我发现了一个很有趣的问题,它们都是负义链上面的基因!
| entrez ID | gene symbol | 正负链 | |
| 911 | CD1C | - | BDCA1|CD1|CD1A|R7 |
| 913 | CD1E | - | CD1A|R2 |
| 909 | CD1A | - | CD1|FCB6|HTA1|R4|T6 |
| 912 | CD1D | - | CD1A|R3 |
| 910 | CD1B | - | CD1|CD1A|R1 |
| 9266 | CYTH2 | - | ARNO|CTS18|CTS18.1|PSCD2|PSCD2L|SEC7L|Sec7p-L|Sec7p-like |
| 30011 | SH3KBP1 | - | CD2BP3|CIN85|GIG10|HSB-1|HSB1|MIG18 |
| 23607 | CD2AP | - | CMS |
| 89886 | SLAMF9 | - | CD2F-10|CD2F10|CD84-H1|CD84H1|SF2001 |
| 10849 | CD3EAP | - | ASE-1|ASE1|CAST|PAF49 |
| 445347 | TARP | - | CD3G|TCRG|TCRGC1|TCRGC2 |
| 915 | CD3D | - | CD3-DELTA|IMD19|T3D |
| 920 | CD4 | - | CD4mut |
| 922 | CD5L | - | AIM|API6|PRO229|SP-ALPHA|Spalpha |
| 925 | CD8A | - | CD8|Leu2|MAL|p32 |
| 927 | CD8BP | - | CD8B2 |
| 54675 | CRLS1 | - | C20orf155|CLS|CLS1|GCD10|dJ967N21.6 |
| 3681 | ITGAD | - | ADB2|CD11D |
| 3683 | ITGAL | - | CD11A|LFA-1|LFA1A |
| 3684 | ITGAM | - | CD11B|CR3A|MAC-1|MAC1A|MO1A|SLEB6 |
| 3687 | ITGAX | - | CD11C|SLEB6 |
| 290 | ANPEP | - | APN|CD13|GP150|LAP1|P150|PEPN |
| 115708 | TRMT61A | - | C14orf172|GCD14|Gcd14p|TRM61|hTRM61 |
| 2526 | FUT4 | - | CD15|ELFT|FCT3A|FUC-TIV|FUTIV|LeX|SSEA-1 |
| 2215 | FCGR3B | - | CD16|CD16b|FCG3|FCGR3|FCR-10|FCRIII|FCRIIIb |
| 4055 | LTBR | - | CD18|D12S370|LT-BETA-R|TNF-R-III|TNFCR|TNFR-RP|TNFR2-RP|TNFR3|TNFRSF3 |
| 930 | CD19 | - | B4|CVID3 |