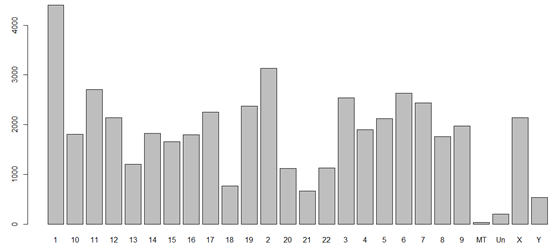
在bioconductor的官网里面可以查到共有111个系列包,基本上跨越了我们常见的物种啦!
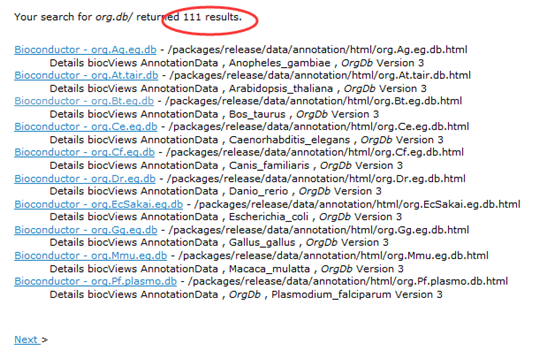
斑马鱼:Bioconductor - org.Dr.eg.db - /packages/release/data/annotation/html/org.Dr.eg.db.html
Details biocViews AnnotationData , Danio_rerio , OrgDb Version 3
拟南芥:Bioconductor - org.At.tair.db - /packages/release/data/annotation/html/org.At.tair.db.html
Details biocViews AnnotationData , Arabidopsis_thaliana , OrgDb Version 3
小鼠:Bioconductor - org.Mm.eg.db - /packages/release/data/annotation/html/org.Mm.eg.db.html
Details biocViews AnnotationData , Mus_musculus , OrgDb , mouseLLMappings Version 3
人类:Bioconductor - org.Hs.eg.db - /packages/release/data/annotation/html/org.Hs.eg.db.html
Details biocViews AnnotationData , Homo_sapiens , OrgDb , humanLLMappings Version 3
对这些系列包的函数都一样,包括以下几个:
columns(x) keytypes(x) keys(x, keytype, ...) select(x, keys, columns, keytype, ...) saveDb(x, file) loadDb(file, dbType, dbPackage, ...)
这些包就是bioconductor已经做好的数据库,我们可以根据定义好的ID号来进行任意的基因转换,现在支持的信息有一下几种!
keytypes(org.Hs.eg.db)
[1] "ENTREZID" "PFAM" "IPI" "PROSITE" "ACCNUM" "ALIAS" "CHR"
[8] "CHRLOC" "CHRLOCEND" "ENZYME" "MAP" "PATH" "PMID" "REFSEQ"
[15] "SYMBOL" "UNIGENE" "ENSEMBL" "ENSEMBLPROT" "ENSEMBLTRANS" "GENENAME" "UNIPROT"
[22] "GO" "EVIDENCE" "ONTOLOGY" "GOALL" "EVIDENCEALL" "ONTOLOGYALL" "OMIM"
[29] "UCSCKG"
这些包的确非常有用,大家可以看我博客里面关于它们的介绍!!!