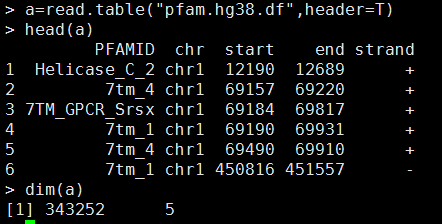
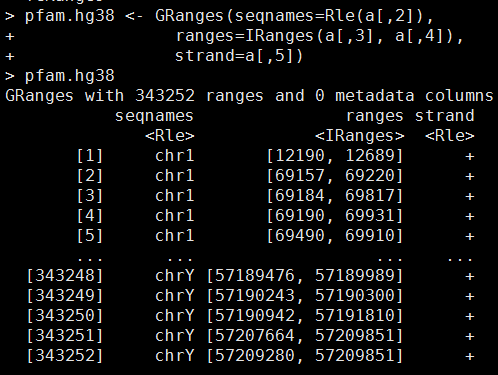
排名不分先后:
推荐宾夕法尼亚州立大学的一个教授Istvan Albert
他写了一本书是: https://www.biostarhandbook.com/
他还可以授予网上课程学位:http://www.personal.psu.edu/iua1/certificate.html
他还推荐了一本R语言书籍:http://onepager.togaware.com/
关注一下华盛顿大学医学院的教授Obi L. Griffith
他的主页:http://www.obigriffith.org/
他的一个比较出名的的贡献是 www.rnaseq.wiki
他在 Biostars bioinformatics forum 非常活跃
关注一下华盛顿大学医学院的教授Malachi Griffith
他的个人主页是:http://www.malachigriffith.org/index.htm
他的github主页是:https://github.com/malachig
WashU TGI Faculty page: Profile
Linked In: Profile
Twitter: Feed
Google Scholar: Citations
Research Gate: Profile
Scopus: Profile
Open Research ID: Profile
Github: Profile
BioStar: Profile
SeqAnswers: Profile
Code Academy: Profile
Iterative Genomics Consulting: Company website
Flickr: Photostream
www.dgidb.org
www.alexaplatform.org
Linked In: Profile
Twitter: Feed
Google Scholar: Citations
Research Gate: Profile
Scopus: Profile
Open Research ID: Profile
Github: Profile
BioStar: Profile
SeqAnswers: Profile
Code Academy: Profile
Iterative Genomics Consulting: Company website
Flickr: Photostream
www.dgidb.org
www.alexaplatform.org
关注一下麦吉尔大学的Pablo Cingolani教授
他是snpeff的作者
他的github是:https://github.com/pcingola
现就职于McGill University
推荐弗吉尼亚大学的stephen教授
他是个人主页:http://stephenturner.us/
他所有公开的ppt : https://speakerdeck.com/stephenturner
stephen教授我要重点提一下,因为他的教育资源特别多。