好的颜值,人人都爱,是你接触有趣的灵魂的敲门砖。单细胞数据分析也是如此,人人都知道需要降维聚类分群。 Continue reading
六
25
好的颜值,人人都爱,是你接触有趣的灵魂的敲门砖。单细胞数据分析也是如此,人人都知道需要降维聚类分群。 Continue reading
传统的bulk转录组测序并没有没落,虽然说大家都在抢单细胞的热点!
最近看到了一个胰腺癌的单细胞文章,公开了其测序数据及表达量矩阵,很方便做图表复现。 Continue reading
前面我们的明码标价之普通转录组上游分析,受到了各大热心粉丝的吐槽,觉得太简单了我们居然还好意思收费。后面我就就加上了稍微有一点难度的《可变剪切》,不过仍然是阻挡不了粉丝无穷无尽的需求,后台有人发给我一个RNA-Seq数据的内含子保留分析需求。
我看了看粉丝发过来的文章,发表于 January 2021, 在CELL杂志的文章《Spliceosome-targeted therapies trigger an antiviral immune response in triple-negative breast cancer》,链接是:https://doi.org/10.1016/j.cell.2020.12.031
这个文章数据比较多:
SUM159 SD6 RNA-Seq #GSE163414
LM2 SD6 RNA-Seq #GSE163411
SUM159 Cytoplasmic RNA-Seq #GSE163232
SUM159 J2 dsRIPseq #GSE163188
Syngeneic model RNA-Seq #GSE163181
可以看到,主要是RNA-Seq数据啦,有两个是普通的细胞系处理前后的表达量差异情况探索,所以出图如下:
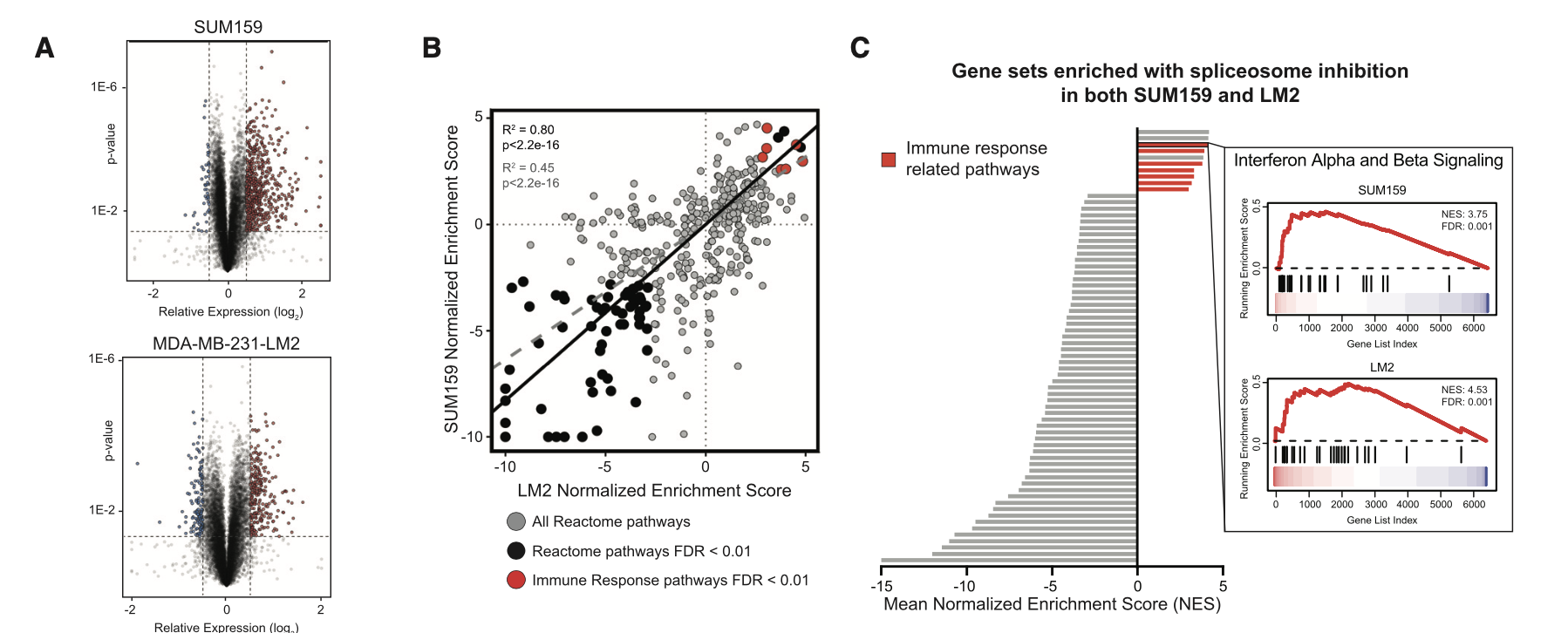
这个已经是超级简单了, 我们的明码标价之转录组常规测序服务(仅需799每个样品) 和 明码标价之普通转录组上游分析 就是对这样的 RNA-Seq拿到了表达量矩阵,然后下游分析也是平淡无奇,仅收费800,代码呢,我也多次分享了,基本上看我六年前的表达芯片的公共数据库挖掘系列推文即可;
这样的分析流程基本上绝大部分粉丝已经是无需委托我们啦,所以粉丝发给我的是 RNA-Seq数据的内含子保留分析需求,步骤如下:
对我们有ngs组学数据分析经验的人来说,其实并不难,无非就是安装几个软件,使用几个包。但对于没有学过编程的纯生物学研究者来说基本上不可能完成,也没有这样的网页工具。
但是呢,这个流程又确实是过于个性化,哪怕对我们来说很简单,也其实是耗费时间和精力需要研发调试的。
如果是TCGA数据库,步骤如下:
一般来说,大家是很难下载TCGA数据库原始fastq文件,这个权限审核比较严厉,不过咱们数据挖掘呢完全没有毕业一直盯着TCGA数据库啊,自己领域的普通RNA-seq肯定也是不少。如果是认真搞科研,你一定会自行调研和阅读文献,找到合适的数据集。
部分粉丝看到这里,可能无法理解RNA-Seq数据的内含子保留分析的意义是什么?
其实就是多了一个维度的指标,来把你的样本分类,分类后就可以找差异。同样的我们可以看这个示例文章,感觉每个样品的IR指标,把病人分成IR高低两个组别,然后走普通的ssGSEA分析,生存分析。
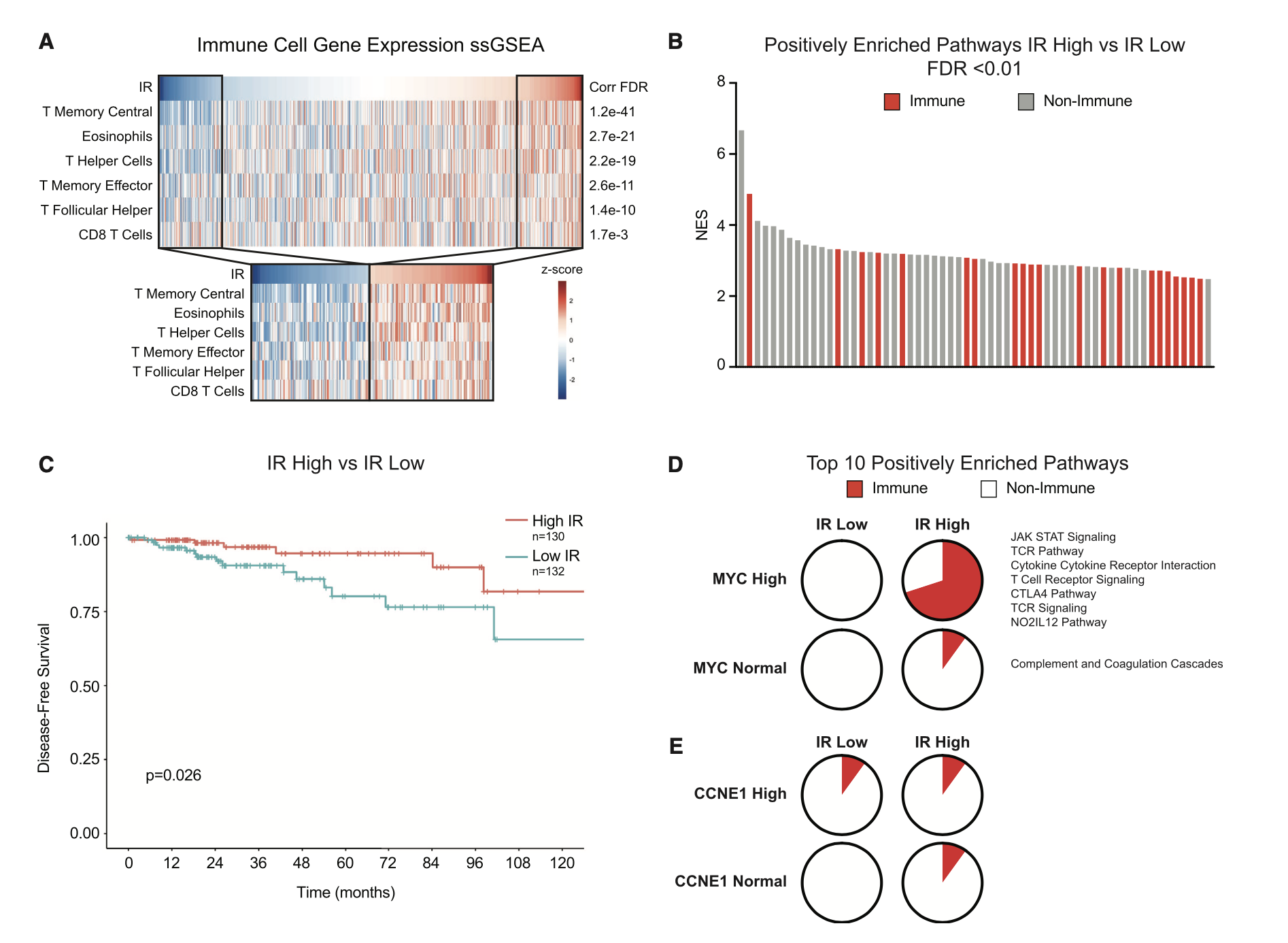
这一套组合拳,大家是不是很眼熟啊?
如果你也想做自己的的RNA-Seq数据的内含子保留分析,赶快联系我们吧,同样的,我们的分析仍然是明码标价,单个RNA-Seq数据的内含子保留分析收费仅需800元,因为是纯粹的基于Linux平台的各种软件脚本,所以提供你全套数据和脚本但是无法保证你能运行成功,因为你不一定有自己的服务器。
最近看到了朋友圈疯转的:[发paper怎么少走弯路?顶刊编辑手把手指点!【文字版】] Continue reading
我在:借鉴escape包的一些可视化GSVA或者ssGSEA结果矩阵的方法 和 对单细胞表达矩阵做gsea分析的两个教程里面提到过,MSigDB(Molecular Signatures Database)数据库中定义了已知的基因集合:http://software.broadinstitute.org/gsea/msigdb 需要注册才能下载。
但是这个GitHub包,ncborcherding/escape文档,在:http://www.bioconductor.org/packages/release/bioc/vignettes/escape/inst/doc/vignette.html 提供了一个封装好的MSigDB数据库信息,其实你仔细看它的文档,它的打包其实是依赖于msigdbr_7.2.1。
MigDB中的全部基因集 都被这个GitHub包,ncborcherding/escape 打包起来了,MSigDB(Molecular Signatures Database)数据库中定义了已知的基因集合:http://software.broadinstitute.org/gsea/msigdb 包括H和C1-C7八个系列(Collection),每个系列分别是:
GS <- getGeneSets(library = "H")
GS
MigDB中的全部基因集 被 构建成为: a list of GSEABase GeneSet objects ,获取 hallmark gene sets (癌症)特征基因集合。
安装方法非常简单:
install.packages("msigdbr")
但是这个msigdbr并没有我想象中的那么大:
Installing package into ‘C:/Users/win10/Documents/R/win-library/4.0’
(as ‘lib’ is unspecified)
试开URL’https://cran.rstudio.com/bin/windows/contrib/4.0/msigdbr_7.2.1.zip'
Content type 'application/zip' length 6737651 bytes (6.4 MB)
downloaded 6.4 MB
package ‘msigdbr’ successfully unpacked and MD5 sums checked
同样的,学习R包,看看文档即可,在: https://cran.r-project.org/web/packages/msigdbr/vignettes/msigdbr-intro.html
Documentation for package ‘msigdbr’ version 7.2.1
DESCRIPTION file.
User guides, package vignettes and other documentation.
Help Pages
msigdbr Retrieve the gene sets data frame
msigdbr_collections List the collections available in the msigdbr package
msigdbr_show_species List the species available in the msigdbr package
msigdbr_species List the species available in the msigdbr package
这些代码使用就明白了,确实没啥好继续讲解的:
library(msigdbr)
# All gene sets in the database can be retrieved without specifying a collection/category.
all_gene_sets = msigdbr(species = "Mus musculus")
head(all_gene_sets)
msigdbr_species()
all_gene_sets = msigdbr(species = "Homo sapiens")
无非就是封装和对象,前面的 escape 包提供了getGeneSets函数,我们的这个msigdbr提供了 msigdbr函数。
B站的10个小时教学视频务必看完,参考 GitHub 仓库存放的相关学习路线指导资料:https://github.com/jmzeng1314/R_bilibili ,可以参考一些优秀笔记,比如https://mubu.com/doc/2KUiSCfVsg
前面我们分享了 跟着Nature Medicine学MeDIP-seq数据分析,数据和代码都是公开,这个2G的压缩包文件,足以学习3个月,写60篇教程。 Continue reading
我们《生信技能树》早期也分享过蛋白质组学数据处理教程,目录如下:
Continue reading
GTEx数据库想必大家并不陌生了,通常我们在挖掘TCGA数据库的时候,会发现该项目纳入的正常组织测序结果是非常少的,也就是说很多病人都不会有他的正常组织的转录组测序结果。 Continue reading
四年前我做了一个单细胞课程,就是对scRNAseq包里面的数据示例的一些处理。 Continue reading
如果你看了我的单细胞转录组数据分析的 基础10讲:

做单细胞数据分析,我们当然希望看到一个清晰的降维聚类分群结果,这样才方便做生物学亚群注释,比如前面的例子:[人人都能学会的单细胞聚类分群注释] Continue reading
看到了发表于2020年8月的一个研究,标题是《Efficient chromatin profiling of H3K4me3 modification in cotton using CUT&Tag》,链接是 https://link.springer.com/article/10.1186/s13007-020-00664-8 Continue reading
众所周知,发布在bioconductor的包主要是生物信息学相关,在官方可以看到其主要是分成3类: Continue reading
绝大部分的肿瘤研究单细胞研究我介绍过 [CNS图表复现08—肿瘤单细胞数据第一次分群通用规则] Continue reading
过去的几年我们一直在强调要想真正入门生物信息学建议务必购买全套书籍,一点一滴攻克计算机基础知识,书单在:什么,生信入门全套书籍仅需160
然后呢,就有非常多的留言期待有电子版PDF分享,但是咱们并不是那样的公众号啊。咱们《生信技能树》,《单细胞天地》等平台,都是生物信息学领域流量天花板,犯不着去使用盗版资源来骗去流量哦!
而且,但凡是跟了我这么多年,学习了一些搜索技巧的,也很容易去搜索到对应的PDF资源啊。不过,我这里有一个更好的推荐,就是微信读书这个APP产品,了解一下,如下所示:

如果你接下来还要问我,什么是微信读书,如何使用,那你可能并不是很适合学习生物信息学!
看了那么多教程,却还是没办法动手分析你的10X单细胞转录组数据,你可能需要这个直播课! Continue reading
也不知道怎么搜索文献就发现了这个有意思的现象:
2017的文章,标题就《Neutrophils dominate the immune cell composition in non-small cell lung cancer》,链接是:https://www.nature.com/articles/ncomms14381 Continue reading
很多人都说传统的bulk转录组测序“廉颇老矣”,急急忙忙转向了单细胞转录组这样的热点技术。 Continue reading
看到一个预印本文章对3种EMT打分算法进行了测评,挺有意思的,标题是:《Comparative study of transcriptomics-based scoring metrics for the epithelial-hybrid-mesenchymal spectrum》,链接在 https://www.biorxiv.org/content/10.1101/2020.01.02.892604v1.full Continue reading