前些天我在 生物学功能注释三板斧,提到了简单的超几何分布检验,复杂一点可以是gsea和gsva,更复杂一点的可以是DoRothEA和PROGENy类似的打分。 Continue reading
二
01
前些天我在 生物学功能注释三板斧,提到了简单的超几何分布检验,复杂一点可以是gsea和gsva,更复杂一点的可以是DoRothEA和PROGENy类似的打分。 Continue reading
有学徒在完成我布置的一个月前( 2023-12-01 )的学徒作业:任意癌症的任意基因突变与否分组后的转录组测序的差异分析的时候,选择了使用TCGAbiolinks包进行TCGA的somatic的突变信息下载,但是他失败了,所以我就帮忙debug了一下。 Continue reading
前面我们在 初试Seurat的V5版本 的推文里面演示了10x单细胞样品的标准3文件的读取,而且在使用Seurat的v5来读取多个10x的单细胞转录组矩阵 的推文里面演示了多个10x单细胞样品的标准3文件的读取。
但是留下来了一个悬念, 就是如果我们的单细胞转录组并不是10x的标准3文件,而是tsv或者csv或者txt等文本文件表达量矩阵信息,就有点麻烦了。接下来我们以2020的文章:《Single-Cell Transcriptome Analysis Reveals Dynamic Cell Populations and Differential Gene Expression Patterns in Control and Aneurysmal Human Aortic Tissue》举例说明,它的数据集是 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE155468 Continue reading
前面我们在 初试Seurat的V5版本 的推文里面演示了文章标题是:《CD36+ cancer-associated fibroblasts provide immunosuppressive microenvironment for hepatocellular carcinoma via secretion of macrophage migration inhibitory factor》,的数据集GSE202642的Seurat的v5读取方式。
前些天我在 生物学功能注释三板斧,提到了简单的超几何分布检验,复杂一点可以是gsea和gsva,更复杂一点的可以是DoRothEA和PROGENy类似的打分。
其中 GO(Gene Ontology)和KEGG(Kyoto Encyclopedia of Genes and Genomes)数据库是两个常用的生物学功能注释数据库,科学家通常是使用来超几何分布检验这个统计学算法做富集分析,即通过比较实际观察到的基因集合(几十个或者几百个)中特定功能或通路的基因数量与随机期望的数量来判断其是否富集。 Continue reading
前些天我在 生物学功能注释三板斧,提到了简单的超几何分布检验,复杂一点可以是gsea和gsva,更复杂一点的可以是DoRothEA和PROGENy类似的打分。
其中 GO(Gene Ontology)和KEGG(Kyoto Encyclopedia of Genes and Genomes)数据库是两个常用的生物学功能注释数据库,但是GO数据库 注释通常包括三个方面的信息:分子功能(Molecular Function)、细胞组分(Cellular Component)和生物过程(Biological Process)。而前面我们演示了:使用topGO增强你的GO数据库注释结果的可视化,是超几何分布检验的结果的可视化,主要是展示GO数据库的有向无环图结构。接下来我们聊聊使用clusterProfiler的GSEA方法针对GO数据库进行注释后的结果的可视化,所以是需要大家自己提前弄清楚GSEA方法和超几何分布检验方法的区别哦! Continue reading
学员做了一个时间序列转录组测序项目,我们帮忙处理了一下,实验设计如下所示:
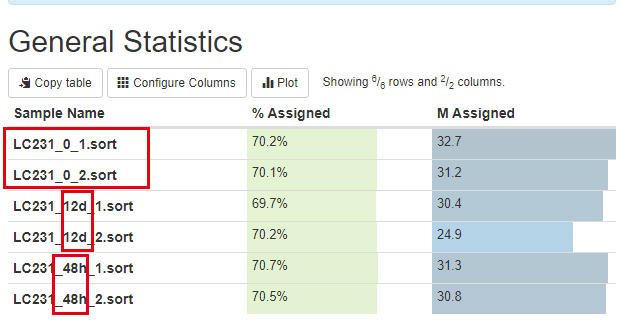
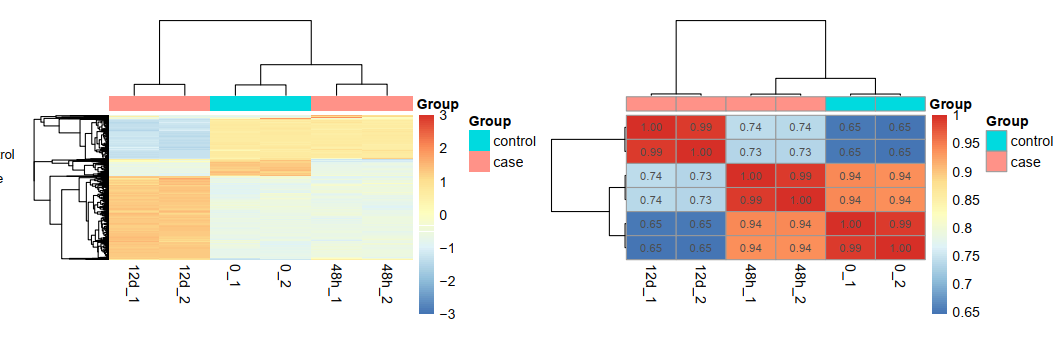
可以看到是12d和48h处理的效果不一样,其中48h的处理跟control更接近,而12d的处理很明显影响很大:
最近很多行业交流群都被因渐冻症而备受关注的原京东副总裁蔡磊的视频刷屏了:
2024年1月27日,京东原副总裁、渐冻症抗争者蔡磊在微博发布消息称,与夫人将再捐助1亿元,用于支持渐冻症的基础研究、药物研发、临床医疗等科研项目。 Continue reading
最近是又搞文章数据分析的图表复现文字版教程,也有视频号的直播互动,详见:殊路同归的关键单细胞亚群鉴定算法,但是阅读量就是起不来。
我可能是悟了,其实大家感兴趣的应该是我本人,过去的七八年间一百多个实习生都有自己的生物信息学笔记公众号都在日更产出知识点,在知识输出这方面我其实并不是我早期的实习生了。而且现在没办法继续培养实习生,我的办公室就空下来了,之前是预留的生信共享办公室出租,也是无人问津,想了想可能是因为基本上很难有人下定决心换一个城市生活和工作。 Continue reading
昨天的视频号直播:踏入生物信息学海洋的必修课是什么,举办方精选出来的读者提问都是很精锐的辩论题,包括:狂敲代码和玩转思路的二选一,临床问题和基础科研孰轻孰重。安排到我和Y叔的一个共同讨论题是:生信分析和实验结果哪个更可靠? Continue reading
因为“众所周知”的原因,我们持续了五六年的实习生培养计划从此落幕了,但是这个事件让小红书走入了我的视线。隔三差五就打开看看里面的关于生物信息学学习和就业市场的真实分享,其中一个小伙伴(河北大学的2019届生信专业本科毕业生)分享了他最近五年的职场进阶之路,就是从研究所再到上市公司再到朝气蓬勃的小公司……
蛮有意思的,让我看到了十年前的我,也是大四就去帝都实习半年那个时候差一点就牺牲在2013的全城雾霾,也是工作三四年就年薪差不多30万了,不过我没有选择继续去涨工资到年薪50万而是蹉跎了七年在尝试科研路。 Continue reading
生物学功能注释是对特定的数量(几十个或者几百个)基因或蛋白的合集的功能进行描述和分类的过程。GO(Gene Ontology)和KEGG(Kyoto Encyclopedia of Genes and Genomes)数据库是两个常用的生物学功能注释数据库,科学家通常是使用来超几何分布检验这个统计学算法做富集分析,即通过比较实际观察到的基因集合(几十个或者几百个)中特定功能或通路的基因数量与随机期望的数量来判断其是否富集。 Continue reading
生物信息学领域涉及到大量的不同种类的数据的分析和处理工作,因此这个领域就必然产生许多不同类型的软件工具,比如处理DNA、RNA、蛋白质序列等不同层面的数据。但是我们这里并不想按照组学种类来对生物信息学软件工具进行分类,因为不同组学经常是有软件是交叉的,比如fastqc软件就可以针对不同ngs组学数据进行质量控制。我这里把生物信息学软件工具按照使用难易程度的大致分成3类: Continue reading
张雪峰最近在直播中又说出了让全网沸腾的“暴论”:“文科都是服务业,什么是服务业?总结成一个字就是‘舔’,就是‘爷我给你笑一个’,‘爷买一号链接吗’”。
张雪峰的意思应该是想说文科专业毕业后不仅仅工资低,而且还不受尊重吧。 Continue reading
最近开始在学习基于R语言的seurat包的单细胞测序数据分析。Jimmy老师给了一个实战分析,在分析过程中逐渐认识seurat包的数据结构。本次推文主要分享一下如何替换seurat对象中的orig.ident为样本名字。 Continue reading
直播展示单细胞降维聚类分群的时候有小伙伴说我们昨天和今天大家结果居然不一样! Continue reading
最近测试了immunedeconv包,首先它在github上面,所以本身就很难安装:
# deconvolution_methods
# https://github.com/omnideconv/immunedeconv
# remotes::install_github("omnideconv/immunedeconv")
library(immunedeconv)
library(tidyverse)
library(tidymodels)
然后我在运行immunedeconv包里面的mcp_counter时候,发现它需要访问一个在github的文本文件:
res.mcp <- deconvolute(expr, 'mcp_counter')
那肯定是会报错:
# genes = read.table(curl:::curl("https://raw.githubusercontent.com/ebecht/MCPcounter/master/Signatures/genes.txt")
# Could not resolve host: raw.githubusercontent.com
然后chatGPT给我了两个解决方案,通过BioinfoArk提供的中国区chatGPT查询: Continue reading
一个月前( 2023-12-01 )的学徒作业:任意癌症的任意基因突变与否分组后的转录组测序的差异分析,陆陆续续收到了一些反馈,有马拉松授课学员的也有学徒实习生的,发现虽然说给大家指明了数据分析结题思路,但大家仍然是千奇百怪的错误。总体上就5个步骤,大家可以错十几处: Continue reading
关键是看如何定义免疫特征,比如kegg数据库里面的就有很多免疫特征相关的功能基因列表,首先它区分成为了如下所示的7个大类: Continue reading
前面我们提到了空间单细胞约等于10x技术,就比较方便理解,虽然说也有其它空间单细胞技术可以产出各式各样的数据。详见:10x的空间单细胞文件格式详解
但是对初学者来说,重要的是如何把不同技术产出的表达量矩阵导入到R或者Python这样的编程语言环境里面。今天我们来介绍的是在R语言里面的最流行的Seurat的单细胞流程,第一步就是理解Seurat的空间单细胞对象结构。值得注意的是我们接下来(2023年12月30日之后)的教程都是基于Seurat的V5版本哦: Continue reading