我首先让chatGPT帮我根据fasta格式的蛋白质序列文件,使用Python语言计算里面的每个蛋白质的序列长度和分子量,它给了描述了解题思路。。。

看起来蛮厉害的,我怀疑它可能是对我的真正的需求不够了解,我其实就是想让他帮忙写代码,就继续追问它: Continue reading
我首先让chatGPT帮我根据fasta格式的蛋白质序列文件,使用Python语言计算里面的每个蛋白质的序列长度和分子量,它给了描述了解题思路。。。
看起来蛮厉害的,我怀疑它可能是对我的真正的需求不够了解,我其实就是想让他帮忙写代码,就继续追问它: Continue reading
如果你去openAI问chatGPT关于孟德尔随机化的流程,它是这样的回答:
是的,我可以为您提供一个孟德尔随机化的流程介绍。
孟德尔随机化是一种常见的生物信息学算法,用于评估基因对生物学过程的影响。该算法通过模拟随机基因重排来评估基因组和外显子组的对生物学目标的影响。
以下是孟德尔随机化的流程: Continue reading
隔三差五就有小伙伴在交流群里面问到他自己在进行CD4和CD8的T细胞细分亚群的时候非常纠结,根本就没办法确定下来到底应该是分多少群以及到底是应该按照什么顺序分。其实很正常,之前我们组建了CD4和CD8的T细胞的交流群就探讨失败,虽然说CD8 T和CD4 T细胞亚群 是一种分类方法,但是各自内部又是可以按照功能进行划分,naive, memory ,effector,cytotoxic,Exhaustion。 Continue reading
最近在交流群看到了一个很有意思的讨论,就是他看到了他们领域的癌症高分文献,发现里面的差异分析结果跟之前的另外一个高分文献里面的基因很不一样,就以为我们生物信息学是万能魔法,可以让任意基因都有差异:
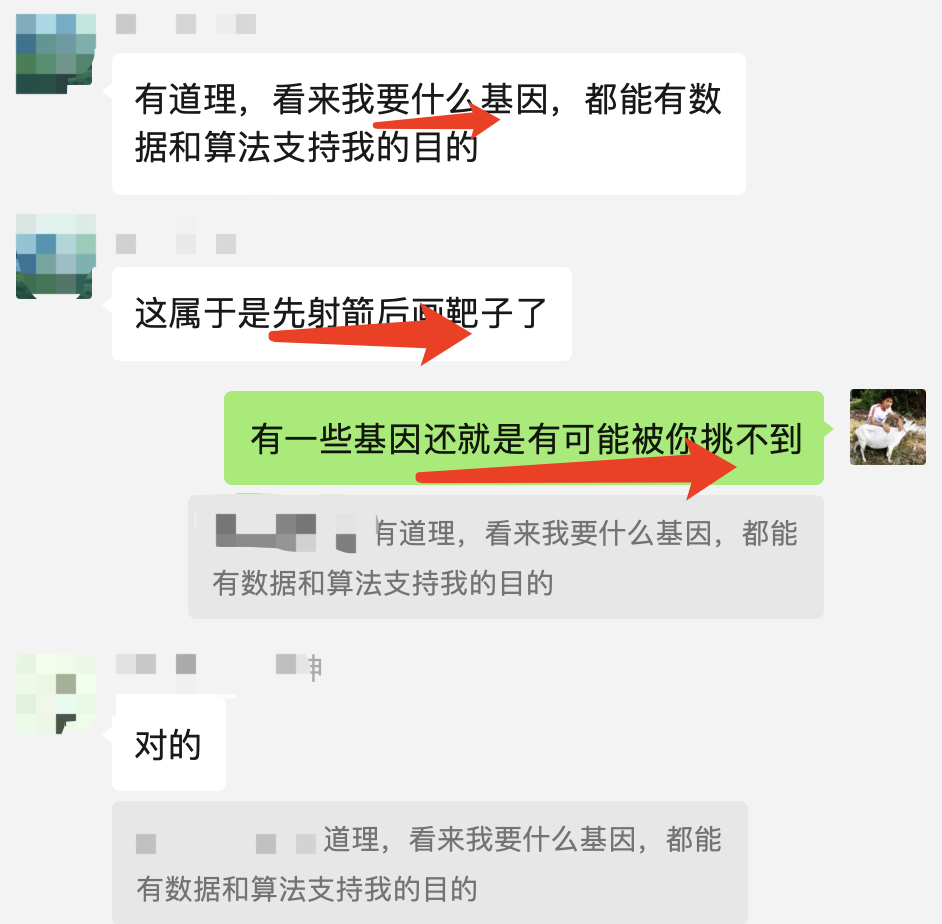 Continue reading
Continue reading
这两天看到了各种交流群以及朋友圈小伙伴都在转发和祝贺张泽民课题组的泛癌层面NK单细胞数据挖掘文章, 我简单看了看,类似的策略的数据挖掘居然一直可以发CNS级别杂志,让人膜拜:
众所周知,单个单细胞的转录组,如果是目前流行的10x这样的单细胞转录组技术,具体到每个细胞层面我们拿到的表达量信息通常是500到2000个基因的范围,也就是说如果我们的参考基因组注释文件里面是2万或者6万基因会出现起码90%的基因是缺失的,这个现象在单细胞转录组领域称作是drop-out (大家可以自行统计一下pbmc3k这个示例数据的drop-out情况 ) Continue reading
基本上每年我们都会在生信技能树等公众号写多个教程分享WGCNA的实战细节,比如: Continue reading
看到了交流群小伙伴分享了一系列数据挖掘文章,都是浙江大学李兰娟院士的学生的成果。其中一个《Characteristic Analysis of Featured Genes Associated with Cholangiocarcinoma Progression》倒是蛮简单的,挑选TCGA数据库里面的 GDC TCGA Bile Duct Cancer (CHOL)&removeHub=https%3A%2F%2Fxena.treehouse.gi.ucsc.edu%3A443) 数据集,然后根据里面的样品的二分类属性(肿瘤样品和正常组织对照)做一个简单的差异分析,然后基于差异分析后的基因列表进行go和kegg的数据库注释,以及使用WGCNA算法构建网络,然后挑选合适的网络看里面的hub基因而已。 Continue reading
前些天,我们的《生信技能树》论坛所在的服务器被我格式化,所以论坛过往七八年的几万篇笔记就灰灰湮灭了。
其中论坛所在服务器背后还有有一个简易的生物信息学书籍,详见:都不知道自己还有一本书,很多人过来询问该如何继续浏览。然后热心的阿越居然帮我找到了存稿,而且帮我重新制作成为了可以被浏览的在线书籍,最新的链接在《生信菜鸟团》博客里面,是: http://bio-info-trainee.com/basic/ (应该算是一个好消息吧!) Continue reading
文章是:《Identification and functional analysis of novel oncogene DDX60L in pancreatic ductal adenocarcinoma》,细读它可以发现两个不同的数据集, (GSE171485 and GSE171486).
学术交流是研究人员与同行、学术界以及社会大众分享他们的研究成果、发现和观点的重要方式。然而,并不是所有的学术交流都必须通过SCI(Science Citation Index)期刊发表文章 :
Zenodo 是一个开放的研究数据存储和共享平台,旨在促进学术界内的数据、出版物和其他研究成果的共享。以下是 Zenodo 平台的主要学术功能:
昨天我们diss了肿瘤外显子技术,很容易设计课题,任意肿瘤对象招募病人集齐样品送给公司进行肿瘤外显子测序即可,公司一般来说也会给出来somatic突变信息,包括SNV和CNV,甚至给出来发表级别的图表。详见:几乎不提供任何有用信息的肿瘤外显子你还做吗 Continue reading
最近看到了一个博客讨论了细胞类型和细胞状态,详见:https://mbernste.github.io/posts/cell_types_cell_states/,里面提到了Svennson 等人(2021)的预印本研究发现,随着研究人员测量更多的细胞,他们往往会发现更多的“细胞类型”。。。
为什么CD4和CD8的T细胞在单细胞转录组水平本来就很难确定亚群和名字,因为绝大部分已经发表的文章对他们的细分都是依据细胞形态,而不是真正的细胞类型。 Continue reading
原文发表在《生信技能树》公众号,详见:https://mp.weixin.qq.com/s/IVAPcGLIZmNoS8YXsCm7Dg
生物信息学是一个跨学科的领域,涉及生物学、计算机科学、数学和统计学等多个领域的知识。因此,入门生物信息学可能会遇到以下一些困难:
前些天有一个人给了我这样的留言,让我我现在很迷茫:
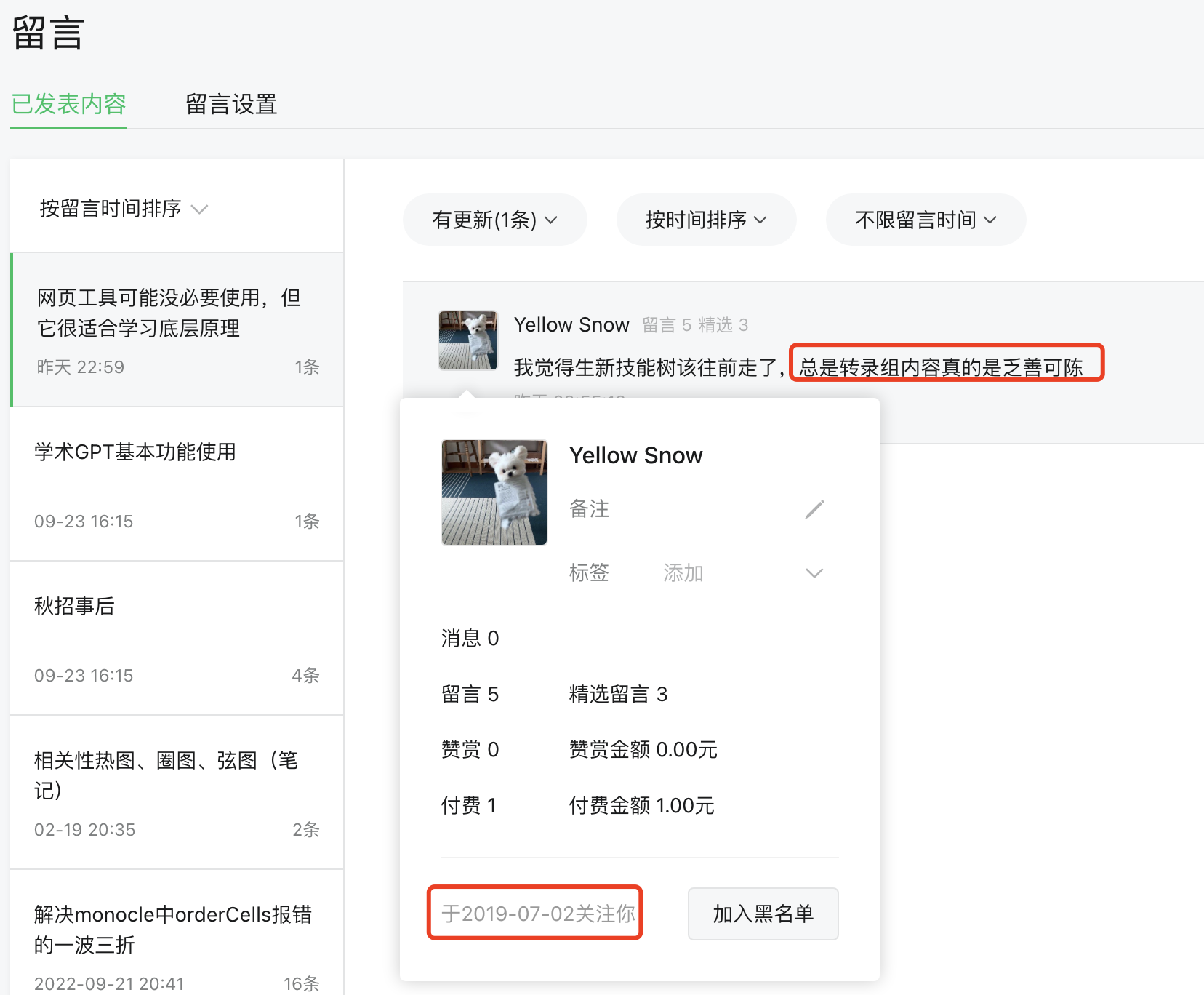
这个人已经是四五年的老粉了,确实,如果他一路看过来,很容易发现我们其实一直是三板斧(编程基础+ngs多组学+单细胞)。很单调,是有点乏善可陈,但是我个人确实是感觉写什么都很难突破过去的自己,这个公众号创作是越来越搞不下去了。。。。 Continue reading
每个人都是从初学者走过来的,我当然是知道纯粹的初学者肯定是没办法凭空去获得如何查看自己的r包安装在电脑什么目录的知识点,但是我觉得这个不应该是在微信交流群里面讨论的,只需要简单的购买书籍去学习最基础r知识点即可。
官网说的很清楚:https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/using/fastq-input
To serve as inputs for cellranger, FASTQ files should conform to the naming conventions of bcl2fastq and mkfastq:
[Sample Name]S1_L00[Lane Number][Read Type]_001.fastq.gz
Where Read Type is one of:
I1: Sample index read (optional)I2: Sample index read (optional)R1: Read 1R2: Read 2 Continue reading 我们《生信菜鸟团》的单细胞周更专辑作者分享过好几次了基础文件读取技巧啦,详见: 读取不同格式的单细胞转录组数据及遇到问题的解决办法。
其中最常见的就是使用Read10X读取3个文件,但是Read10X读取3个文件还得注意版本,而且必须保证3个文件名字完全一样,要么是 Continue reading
最近刷到了生物信息学领域著名期刊NAR的最新文章:《NORMSEQ – a tool for evaluation, selection and visualization of RNA-Seq normalization methods》是一个网页工具,NORMSEQ,它介绍了一些转录组测序表达量矩阵的归一化标准化方法学而且提供了一个在线网页工具给大家使用。
之所以注意到它,是因为NORMSEQ的流程图画的很吸引眼球: Continue reading