前面我在 十多万个细胞数量就顶不住了吗提到了一个关键的转换, 稀疏矩阵:
ct <- as.matrix(ct, type="dgCMatrix")
马上交流群就有小伙伴反馈了同款的bug,是二十多万个细胞的单细胞数据集,但是他自己就算是转换成为稀疏矩阵也没有解决问题。。。
ct=data.table::fread('GSE183852_Integrated_Counts.csv.gz',
data.table = F)
ct[1:4,1:4]
tail(ct[ ,1:4])
rownames(ct)=ct[,1]
ct=ct[,-1]
tmp <- as.matrix(ct, type="dgCMatrix")
sce.all=CreateSeuratObject(counts = tmp )
如下所示:
> ct[1:4,1:4]
gene H_ZC-11-292_TAAGTGCAGCAGGTCA H_ZC-11-292_ACAGCCGGTCATACTG
1 RP11-34P13.3 0 0
2 FAM138A 0 0
3 OR4F5 0 0
4 RP11-34P13.7 0 0
> tail(ct[ ,1:4])
gene H_ZC-11-292_TAAGTGCAGCAGGTCA
45063 AC136352.3 0
45064 AC136352.2 0
45065 AC171558.3 0
45066 BX004987.1 0
45067 AC145212.1 0
45068 MAFIP 0
Warning: Data is of class matrix. Coercing to dgCMatrix.
Error: vector memory exhausted (limit reached?)
我也确实是在我们的共享服务器(2024的共享服务器交个朋友福利价仍然是800)测试了,发现并没有问题。就继续沟通,发现交流群的小伙伴其实还没有使用我们的超大内存服务器,仅仅是在自己的电脑里面跑这个代码,所以失败完全是理所当然啊!!!
但是,问题还是得为他解决,上面的GSE183852数据集是有接近27万个细胞,所以绝大部分人普通电脑很难跑通这个环节,很正常。
我做了一个简单的磁盘中间环节避免这个内存bug,如下所示:
lapply(0:8, function(i){
print(i)
kp= seq(1,ncol(ct)) %in% seq(i*30000 ,(i+1)*30000)
print(table(kp))
tmp= ct[,kp]
tmp=as.matrix( tmp ,
type="dgCMatrix")
save(tmp,file = paste(i,'tmp.Rdata'))
})
也就是说,GSE183852数据集是有接近27万个细胞,我把它拆分成为了9个独立的3万个细胞的表达量矩阵文件并且存储在硬盘里面。
然后就可以关闭R后重新打开,进行批量读取:
library(Seurat)
sceList = lapply(0:8, function(i){
print(i)
load(file = paste(i,'tmp.Rdata'))
print(dim(tmp))
sce =CreateSeuratObject(counts = tmp,
min.cells = 5,
min.features = 300 )
print(sce)
return(sce)
})
sce.all=merge(x=sceList[[1]],
y=sceList[ -1 ] )
names(sce.all@assays$RNA@layers)
sce.all[["RNA"]]$counts
LayerData(sce.all, assay = "RNA", layer = "counts")
sce.all <- JoinLayers(sce.all)
dim(sce.all[["RNA"]]$counts )
当然了,如果是你有服务器(2024的共享服务器交个朋友福利价仍然是800),就没必要上面的这样的如此曲折的分析了。
接下来就可以进行常规的降维聚类分群哈, 如下所示的初步结果:
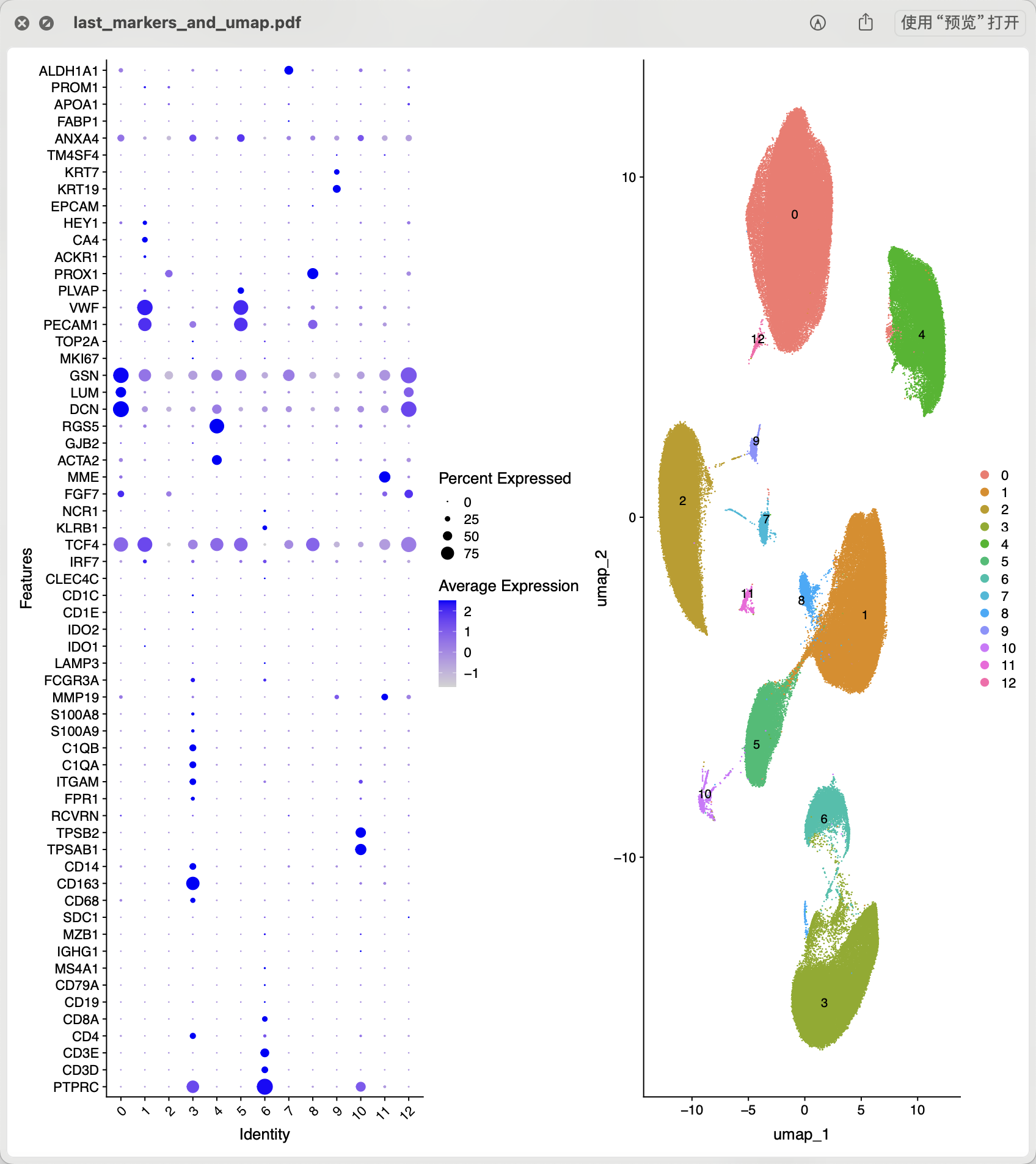
基本上可以看到下面的这些亚群是可以手动命名的:
celltype[celltype$ClusterID %in% c( 6 ),2]='lymphocytes'
celltype[celltype$ClusterID %in% c( 3 ),2]='myeloids'
celltype[celltype$ClusterID %in% c( 1,5 ),2]='endo'
celltype[celltype$ClusterID %in% c( 8 ),2]='L-endo'
celltype[celltype$ClusterID %in% c( 9 ),2]='epi'
celltype[celltype$ClusterID %in% c( 0 ),2]='fibro'
celltype[celltype$ClusterID %in% c( 4 ),2]='SMC'
celltype[celltype$ClusterID %in% c( 10 ),2]='Mast'
celltype[celltype$ClusterID %in% c( 12 ),2]='double'
然后需要看文章里面的基因列表:
补充进行可视化:
cg='RYR2 MYH11 DCN VWF CCL21 NRXN1 HAS1 KIT MZB1 PLIN1 C1QC KCNJ8 LEPR CD3E'
gene_list=trimws(strsplit(cg,' ')[[1]])
gene_list
p2 = DotPlot( sce.all.int, features = gene_list,
group.by = 'RNA_snn_res.0.1') +
theme(axis.text.x = element_text(angle = 45, vjust = 0.5, hjust=0.5))
p2
ggsave('heart-0.1.pdf',width=8)
就可以继续给名字
celltype[celltype$ClusterID %in% c(11 ),2]='Adipo'
celltype[celltype$ClusterID %in% c( 7 ),2]='Neuron'
celltype[celltype$ClusterID %in% c( 2 ),2]='CM'
最后的效果,我感觉我做的比文章要好一点,哈哈哈,如下所示:

学徒作业
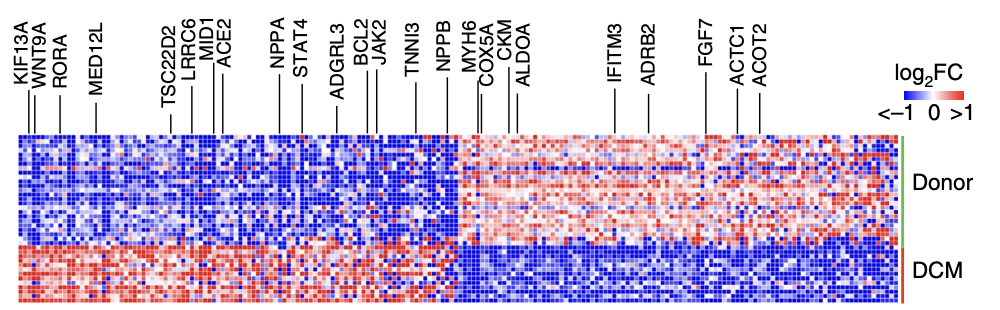
首先呢,完成上面的GSE183852数据集 接近27万个细胞的降维聚类分群和命名,然后读一下文章:《Single-cell transcriptomics reveals cell-type- specific diversification in human heart failure》,然后做一个简单的差异分析即可:
Differential expression analy- sis by pseudobulk and single-cell approaches across disease state revealed a large number of genes significantly upregulated (NPPA, NPPB, ACE2 and KIF13A) and downregulated (MYH6, ADRB2 and CKM) in DCM samples compared to non-diseased donors
并且绘制如下所示的差异基因的热图: