R studio公司毕竟是商业化公司,在R语言推广方面做得很棒。网站什么总共有9个cheatsheet,R语言入门完全可以把这个当做笔记,写代码随时查用!
我批量下载了所有,但是想打印的时候,发现挺麻烦的,因为我不知道批量打印的方法,索性我还是半个程序猿,所以搜索了一下批量合并pdf的方法,这样就可以批量打印了,也方便传输这个文件。
其实如果在linux系统里面,一般都会自带pdf toolkit工具,里面有命令可以合并PDF文档。 Continue reading
R studio公司毕竟是商业化公司,在R语言推广方面做得很棒。网站什么总共有9个cheatsheet,R语言入门完全可以把这个当做笔记,写代码随时查用!
我批量下载了所有,但是想打印的时候,发现挺麻烦的,因为我不知道批量打印的方法,索性我还是半个程序猿,所以搜索了一下批量合并pdf的方法,这样就可以批量打印了,也方便传输这个文件。
其实如果在linux系统里面,一般都会自带pdf toolkit工具,里面有命令可以合并PDF文档。 Continue reading
我前面写到了生信分析人员如何入门linux和perl,后面还会写R和python的总结,但是在这中间我想插入一个脚本实战指南。其实在我前两篇日志里面也重点提到了学习编程语言最重要的就是实战了,也点出了几个关键词。在实际生物信息学数据处理中应用perl和linux,可以借鉴EMBOSS软件套件,fastx-toolkit等基础软件,实现并且模仿该软件的功能。尤其是SMS2/exonerate/里面的一些常见功能,还有DNA2.0 Bioinformatics Toolbox的一些工具。如果你这些名词不懂,请赶快谷歌!!! 它们做了什么,输入文件是什么,输出文件是什么,你都可以用脚本实现!
pwd/ls/cd/mv/rm/cp/mkdir/rmdir/man/locate/head/tail/less/morecut/paste/join/sort/uniq/wc/cat/diff/cmp/aliaswget/ssh/scp/curl/ftp/lftp/mysql/
软硬链接区别文本编辑,文件权限设置打包压缩解压操作(tar/gzip/bzip/ x-j x-c vf)软件的快捷方式如何实现?软件如何安装(源码软件,二进制可执行软件,perl/R/python/java软件)软件版本如何管理,各种编程语言环境如何管理,模块如何管理?(尤其是大部分没有root权限)
二是shell脚本,类似于windows的bat批处理文件
三是高级运维技巧
呀,这是去年(2015)蹭的一个论坛,不记得是第几届了,反正是生物谷举办的,他们搞论坛已经成为一个产业了,非常挣钱的!我那时候还很认真的做了笔记,现在回过头来看看,他们好像讲的都很有道理,虽然我直到现在也用不上,不过我丝毫不担心。我一直拼命的学习各种知识,就是因为有着坚定的信念,所学的一切终将会有一天对我的人生有所帮助。
R与ASReml-R统计分析教程(林元震)中国林业出版社
1-3章简单介绍了R的基本语法,然后第4章着重讲了各种统计方法,第5章讲R的绘图,最后一张讲ASReml-R这个包
语法重点:
1,install.packages(),library(),help(),example(),demo(),length(),attribute(),class(),mode(),dim(),names(),str(),head(),
tail()
2,rep,seq,paste,array,matrix,data.frame,list,c(),factor(),
3,缺失值处理(na.omit,na.rm=T),类型转换(as.numeric(),as.character(),as.factor(),as.logical())
这个软件在TCGA计划里面被频繁使用者,用这个软件的目的很简单,就是你研究了很多癌症样本,通过芯片得到了每个样本的拷贝数变化信息,芯片结果一般是segment结果,可以解释为CNV区域,需要用GISTIC把样本综合起来分析,寻找somatic的CNV,并且注释基因信息。
有两个难点,一是在linux下面安装matlab工作环境,二是如何制作输入文件。
| Human Pseudogene Annotation |
GENCODE Annotation- Data: The current human pseudogene annotation is in GENCODE 21. . - Description: The GENCODE annotation of pseudogenes contains models that have been created by the Human and Vertebrate Analysis and Annotation (HAVANA) team, an expert manual annotation team at the Wellcome Trust Sanger Institute. This is informed by, and checked against, computational pseudogene predictions by thePseudoPipe and RetroFinder pipelines. PseudoPipe Output- Data: The current PseudoPipe results are on Ensembl genome release 79. . - Description: Genome-wide human pseudogene annotation predicted by PseudoPipe. PseudoPipe is a homology-based computational pipeline that searches a mammalian genome and identifies pseudogene sequences. - Reference: Other Human Pseudogene Sets- Data: . - Description: Archived pseudogene annotation on previous human genome releases from PseudoPipe. Genome-wide annotation or specific subset. |
如果你研究癌症,那么TCGA计划的如此丰富的公共数据你肯定不能错过,一般人只能获取到level3的数据,当然,其实一般人也没办法使用level1和level2的数据,毕竟近万个癌症样本的原始测序数据,还是很恐怖的,而且我们拿到原始数据,再重新跑pipeline,其实并不一定比人家TCGA本身分析的要好,所以我们直接拿到分析结果,就足够啦!
mutation signature这个概念提出来还不久,我看了看文献,最早见于2013年的一篇nature文章,主要是用来描述癌症患者的somatic mutation情况的。
首先要自己分析癌症样本数据,拿到somatic mutation,TCGA计划发展到现在已经有非常多的somatic mutation结果啦,大家可以自行选择感兴趣的癌症数据拿来研究,解析一下mutation signature 。
我这里给大家推荐一个工具,是R语言的Bioconductor系列包中的一个,SomaticSignatures
其实它的说明书写的非常详细了已经,如果你理解了mutation signature的概念,很容易用那个包,其实你自己写一个脚本也是非常任意的,就是根据mutation的位置在基因组中找到它的前后一个碱基,然后组成三碱基突变模式,最后统计一下那96种突变模式的分布状况!
我这里简单讲一讲这个包如何用吧!
首先下载并加载几个必须的包:
然后根据突变数据做好一个GRanges对象,这个可以看我以前的博客
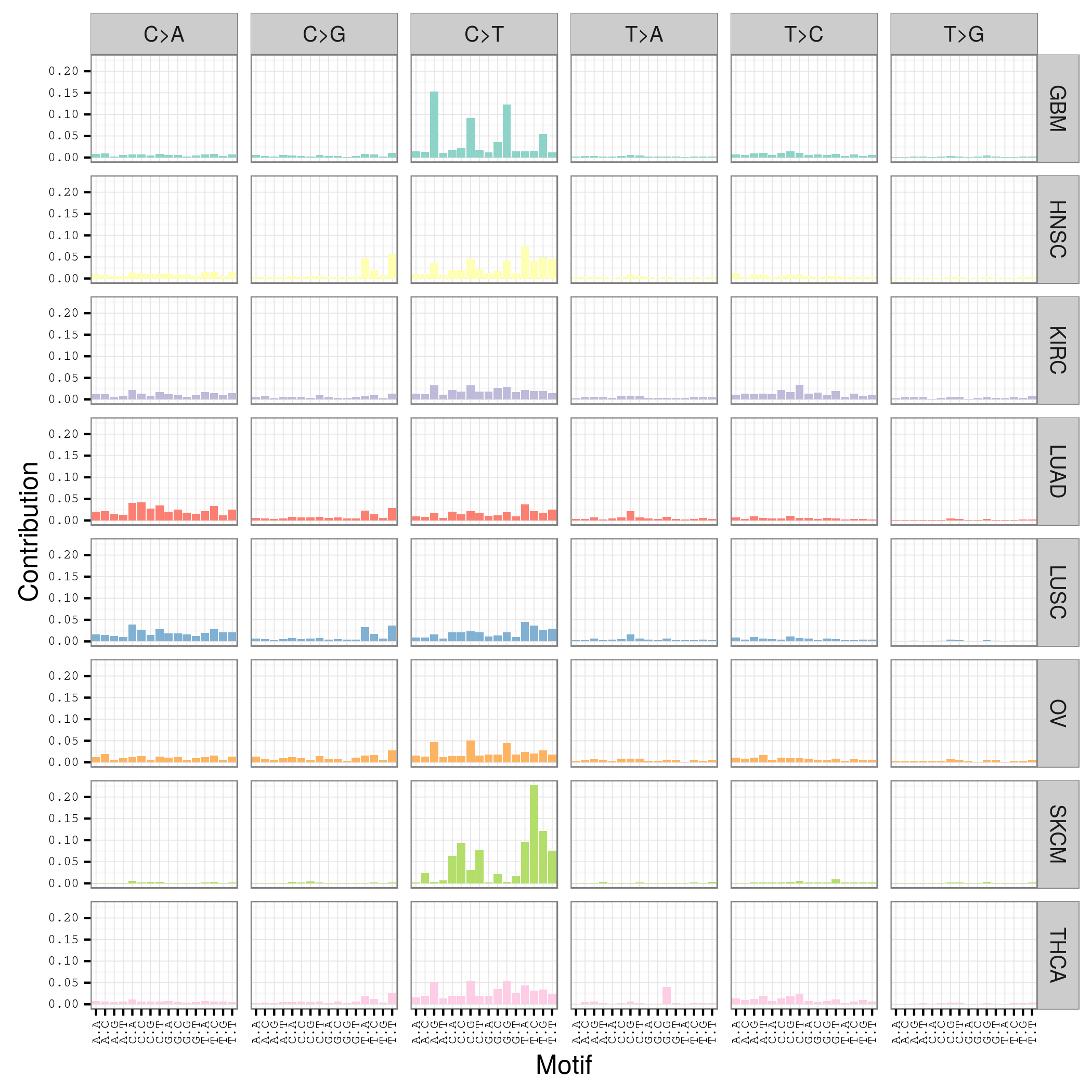
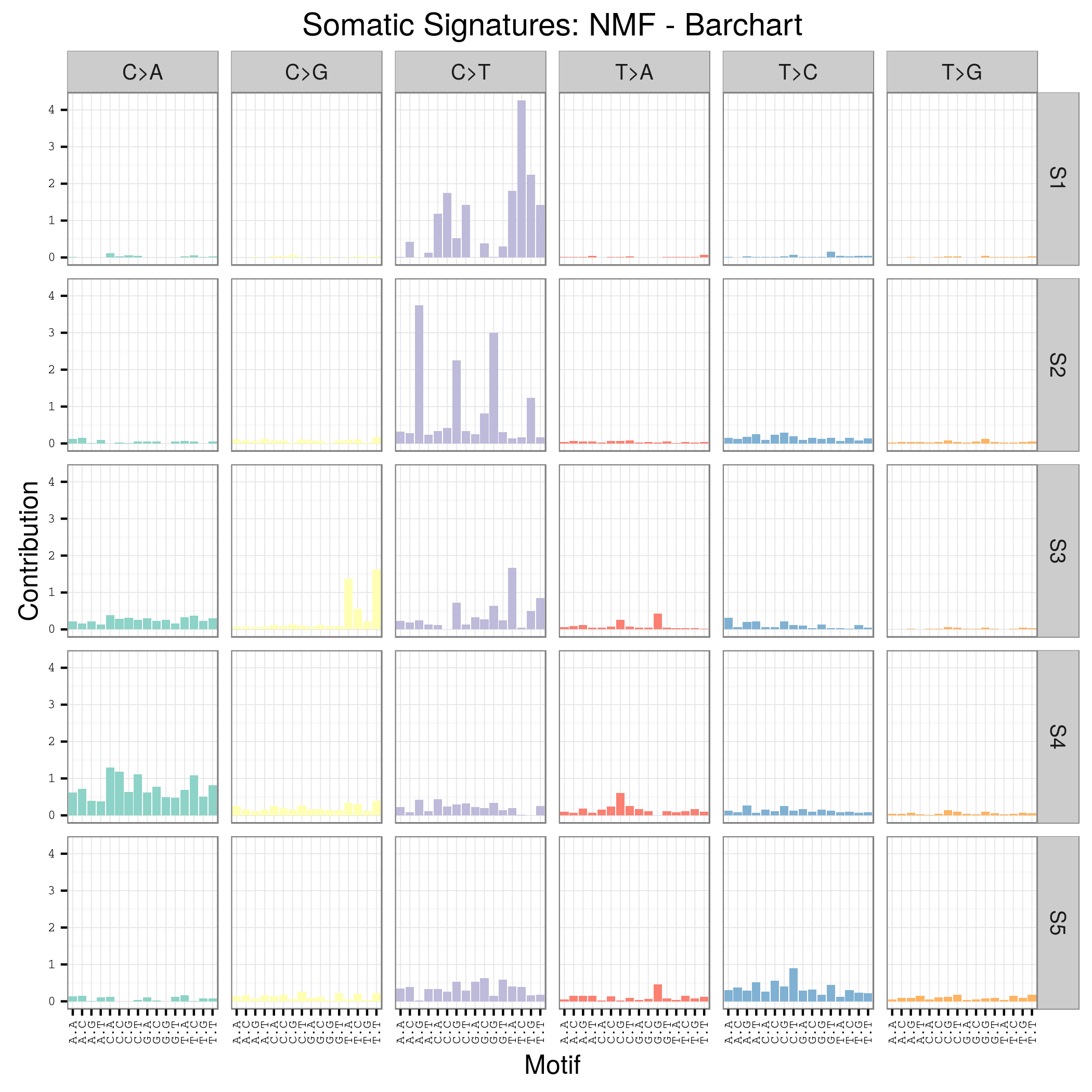
这也是对TCGA数据的深度挖掘,从而提出的一个统计学概念。文章研究了30种癌症,发现21种不同的mutation signature。如果理解了,就会发现这个其实蛮简单的,他们并不重新测序,只是拿已经有了的TCGA数据进行分析,而且居然是发表在nature上面!
研究了4,938,362 mutations from 7,042 cancers样本,突变频谱的概念只是针对于somatic 的mutation。一般是对癌症病人的肿瘤组织和癌旁组织配对测序,过滤得到的somatic mutation,一般一个样本也就几百个somatic 的mutation。
paper链接是:http://www.nature.com/nature/journal/v500/n7463/full/nature12477.html
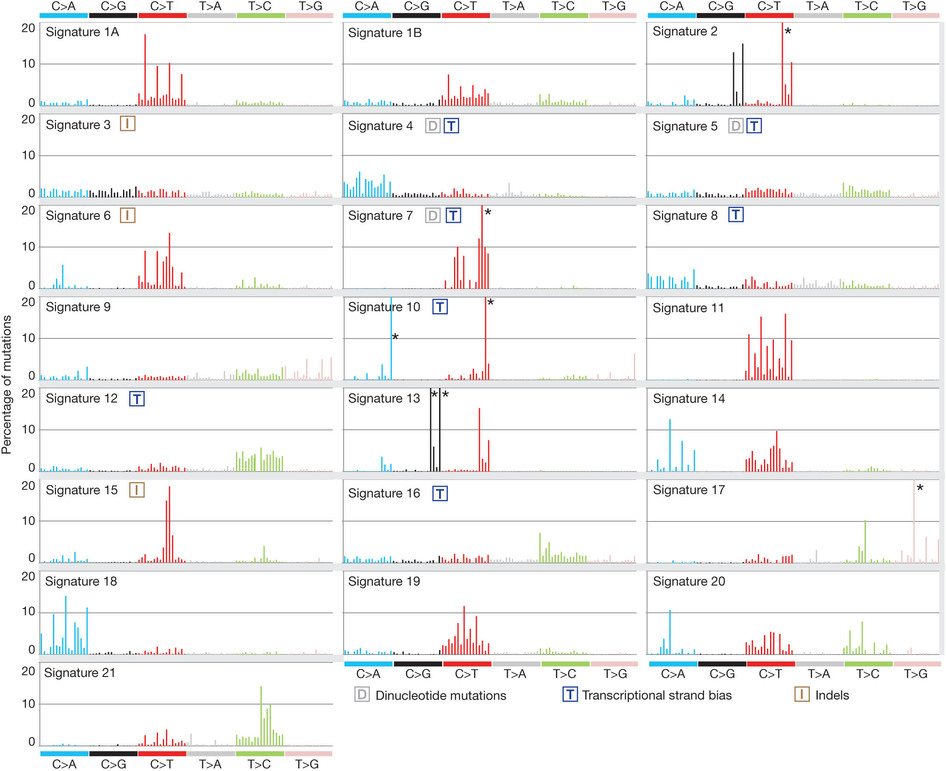
也不知道是什么原因,对国产软件总是提不起兴趣,所以尽管SOAP系列都已经发展到了十几个软件了,我依然没有去试用一下。
# download a test reference genome (TAIR9 Chromosome 1)
wgethttp://biocluster.ucr.edu/~tbackman/query.fastq# download some test Illumina reads from Arabidopsis
运行命令:
2bwt-builder genome.fasta
# create binary of reference genome
soap -a query.fastq -D genome.fasta.index -o output.soap
# align query to genome and store output
结果解读:
tar jxvf software.tar.bz2cd software./configure --prefix=$pathmakemake test
# download a test reference genome (TAIR9 Chromosome 1)
wgethttp://biocluster.ucr.edu/~tbackman/query.fastq# download some test Illumina reads from Arabidopsis
运行命令:
maq # inspect command line options
maq fasta2bfa genome.fasta genome.bfa
# create binary of reference genome
maq fastq2bfq query.fastq readBinary.bfq
# create a binary of dataset
maq match out.map genome.bfa readBinary.bfq
# align query to genome and store output
结果解读:
out.map肯定不是sam格式的。哈哈,这个软件我无法安装,换了好几系统也没成功,如果是太老了,很多库文件却是。我也懒得去解决了。这种报错,对我这样的非计算机专业来说,简直是天书!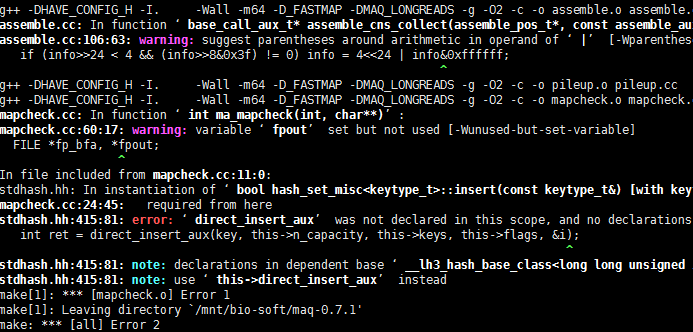
本来搞差异分析的工具和包就一大堆了,而且limma那个包已经非常完善了,我是不准备再讲这个的,正好有个同学问了一下这个包,我就随手测试了一下,顺便看看它跟limma有什么差异没有!手痒了就记录了测试流程!
学习一个包其实非常简单,就是找到包的官网看看说明书即可!说明书链接
第一次听说这个软件,是一个香港朋友推荐的:http://davetang.org/muse/2016/01/13/getting-started-with-gemini/ 他写的很棒,但是我当初以为是一个类似于SQLite的数据库浏览模式,所以没在意。实际上,我现在仍然觉得这个软件没什么用!
软件官网有详细的介绍:https://gemini.readthedocs.io/en/latest/
而且提供丰富的教程:
We recommend that you follow these tutorials in order, as they introduce concepts that build upon one another.
软件本身并不提供注释,虽然它的功能的确包括注释,号称可以利用(ENCODE tracks, UCSC tracks, OMIM, dbSNP, KEGG, and HPRD.)对你的突变位点注释,比如你输入1 861389 . C T ,它告诉你这个突变发生在哪个基因,对蛋白改变如何?是否会产生某些疾病?
虽然它本身没有注释功能,但是它会调用snpEFF或者VEP进行注释,你需要自己先学习它们。
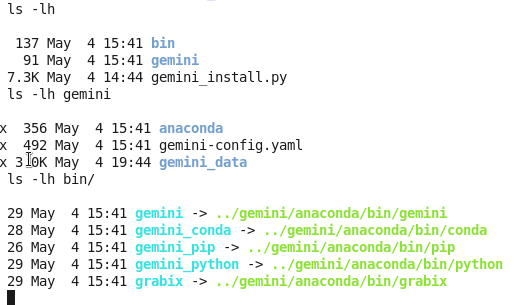
GEMINI是用python写的,有一个小脚本可以自动完成安装过程:
7.3K May 4 14:44 gemini_install.py
下载这个脚本,然后安装即可
wget https://github.com/arq5x/gemini/raw/master/gemini/scripts/gemini_install.py
python gemini_install.py $tools $data
PATH=$tools/bin:$data/anaconda/bin:$PATH
where $tools and $data are paths writable on your system.
我把$tools用的就是当前文件夹,$data也是当前文件夹下面的gemini文件夹。
这样就会在当前文件夹下面生成两个文件夹,bin是存储程序,gemini是存储数据用的,而且注意要把bin目录的全路径添加到环境变量!
我们可以直接下载软件作者提供的测试数据
首先是22号染色体的所有突变位点经过WEP注释的文件
然后是一个三口直接的突变ped格式数据
数据存放在亚马逊云,所有的教程pdf也在
http://s3.amazonaws.com/gemini-tutorials
如果是你自己的vcf文件,需要自己用VEP注释一下


产生是chr22.db就是一个数据库格式的文件,但是需要用gemini 来进行查询,个人认为,并没有多大意思!
你只要熟悉mySQL等SQL语言,完全可以自己来!
VEP是国际三大数据库之一的ENSEMBL提供的,也是非常主流和方便,但它是基于perl语言的,所以在模块方面可能会有点烦人。跟snpEFF一样,也是对遗传变异信息提供更具体的注释,而不仅仅是基于位点区域和基因。如果你熟悉外显子联盟这个数据库EXAC(ExAC.r0.3.sites.vep.vcf.gz),你可以下载它所有的突变记录数据,看看它对每个变异位点到底注释了些什么,它就是典型的用VEP来注释的。 Continue reading
这个软件比较重要,尤其是对做遗传变异相关研究的,很多人做完了snp-calling后喜欢用ANNOVAR来进行注释,但是那个注释还是相对比较简单,只能得到该突变位点在基因的哪个区域,那个基因这样的信息,如果想了解更具体一点,就需要更加功能化的软件了,snpEFF就是其中的佼佼者,而且是java平台软件,非常容易使用!而且它的手册写的非常详细:http://snpeff.sourceforge.net/SnpEff_manual.html Continue reading