string数据库是PPI领域里面最完备已经最受欢迎的数据库了。如果直接在谷歌里面搜索PPI,映入眼帘就是string的官网,它们的主页现在是html5啦,比较精美: http://string-db.org/
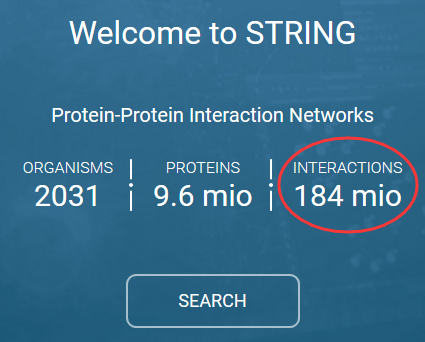
写的很霸气,近两亿的记录,不过一般大家只会关心一个物种,比如人,其实还不到一千万!
我们直接进入下载界面,找到人类的数据,人类的物种ID是9606.
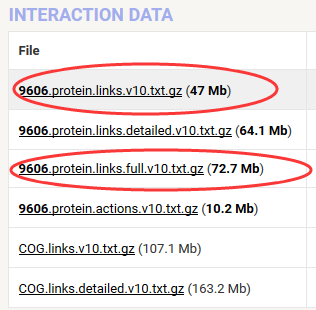
需要一定许可才能下载完整版本,我这里测试最上面那个公开版本数据!
数据很简单,就是protein+protein+score,共八百多万行记录,记录着string数据库搜集的所有可能以及可信的蛋白相互作用!但是它的蛋白ID是ENSEMBL的ID,所以需要转换成基因的ID,才能被大多数人使用,因为大家的研究单位一般是基因,所以蛋白相互作用略等于基因相互作用。
基因ID转换,我推荐用org.Hs.eg.db这个R的包,很容易就可以实现的!
> tmp=toTable(org.Hs.egENSEMBLPROT) > dim(tmp) [1] 110916 2 > head(tmp) gene_id prot_id 1 1 ENSP00000263100 2 1 ENSP00000470909 3 2 ENSP00000443302 4 2 ENSP00000323929 5 2 ENSP00000438599 6 2 ENSP00000445717 |
|
|
有约500多个蛋白ID是无法转换成对应的基因的,这个很正常,毕竟这种ID本来就不稳定,很多用着用着就失效了!
转换好之后就可以上传到数据库啦,然后可以供其它可视化或者分析程序使用!