因为视频教程架构出了点问题,所有不会更新了。现在公布所有下载链接,大家不需要去优酷观看了,太不清晰了,而且还有广告!
视频百度云盘链接:http://pan.baidu.com/s/1jIvwRD8
下面是以前写的大纲,虽然我不做视频了,但是大家可以按照下面的大纲来自学,希望能对你有所帮助!!!
因为视频教程架构出了点问题,所有不会更新了。现在公布所有下载链接,大家不需要去优酷观看了,太不清晰了,而且还有广告!
视频百度云盘链接:http://pan.baidu.com/s/1jIvwRD8
下面是以前写的大纲,虽然我不做视频了,但是大家可以按照下面的大纲来自学,希望能对你有所帮助!!!
这个程序是我在VirusFinder里面发现的,大家可以自行搜索它!
非常好用,建议大家写程序都可以加上这个!
print "\nChecking Java version...\n\n";my $ret = `java -version 2>&1`;print "$ret\n";if (index($ret, '1.6') == -1) {printf "Warning: The tool Trinity of the Broad Institute may require Java 1.6.\n\n";}print "\nChecking SAMtools...\n\n";$ret = `which samtools 2>&1`;if (index($ret, 'no samtools') == -1) {printf "%-30s\tOK\n\n", 'SAMtools';}else{printf "%-30s\tnot found\n\n", 'SAMtools';}my @required_modules = ("Bio::DB::Sam","Bio::DB::Sam::Constants","Bio::SeqIO","Bio::SearchIO","Carp","Config::General","Cwd","Data::Dumper","English","File::Basename","File::Copy","File::Path","File::Spec","File::Temp","FindBin","Getopt::Std","Getopt::Long","IO::Handle","List::MoreUtils","Pod::Usage","threads");print "\nChecking CPAN modules required by VirusFinder...\n\n";my $count = 0;for my $module (@required_modules){eval("use $module");if ($@) {printf "%-30s\tFailed\n", $module;$count++;}else {printf "%-30s\tOK\n", $module;}}if ($count==1){print "\n\nOne module may not be installed properly.\n\n";}elsif ($count > 1){print "\n\n$count modules may not be installed properly.\n\n";}else{print "\n\nAll CPAN modules checked!\n\n";}
其实可以手动下载local::lib, 这个perl模块,然后自己安装在指定目录,也是能解决模块的问题!
下载之后解压,进入:
$ perl Makefile.PL --bootstrap=~/.perl ##这里设置你想把模块放置的目录
$ make test && make install
$ echo 'eval $(perl -I$HOME/.perl/lib/perl5 -Mlocal::lib=$HOME/.perl)' >> ~/.bashrc
等待几个小时即可!!!
添加好环境变量之后,就可以用
perl -MCPAN -Mlocal::lib -e 'CPAN::install(LWP)'
其实你也可以直接打开 ~/.bashrc,然后写入下面的内容
PERL5LIB=$PERL5LIB:/PATH_WHERE_YOU_PUT_THE_PACKAGE/source/bin/perl_module; export PERL5LIB
可以把perl模块安装在任何地方,然后通过这种方式去把模块加载到你的perl程序!
不管别人怎么说,反正我是非常喜欢perl语言的!
也会继续学习,以前写过不少perl模块的博客,发现有点乱,正好最近看到了关于local::lib这个模块。
居然是用来解决没有root权限的用户安装,perl模块问题的!
首先说一下,如果是root用户,模块其实没有问题,直接用cpan下载器,几乎能解决所有的模块下载安装问题!
但是如果是非root用户,那么就麻烦了,很难用自动的cpan下载器,这样只能下载模块源码,然后编译,但是编译有个问题,很多模块居然是依赖于其它模块的,你的不停地下载其它依赖模块,最后才能解决,特别麻烦
但是,只需要运行下面的代码:
wget -O- http://cpanmin.us | perl - -l ~/perl5 App::cpanminus local::lib eval `perl -I ~/perl5/lib/perl5 -Mlocal::lib` echo 'eval `perl -I ~/perl5/lib/perl5 -Mlocal::lib`' >> ~/.profile echo 'export MANPATH=$HOME/perl5/man:$MANPATH' >> ~/.profile
就能拥有一个私人的cpan下载器,~/.profile可能需要更改为.bash_profile, .bashrc, etc等等,取决于你的linux系统!
然后你直接运行cpanm Module::Name,就跟root用户一样的可以下载模块啦!
或者用下面的方式在shell里面安装模块,其中ext是模块的安装目录,可以修改
perl -MTime::HiRes -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Time::HiRes;
perl -MFile::Path -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext File::Path;
perl -MFile::Basename -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext File::Basename;
perl -MFile::Copy -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext File::Copy;
perl -MIO::Handle -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext IO::Handle;
perl -MYAML::XS -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext YAML::XS;
perl -MYAML -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext YAML;
perl -MXML::Simple -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext XML::Simple;
perl -MStorable -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Storable;
perl -MStatistics::Descriptive -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Statistics::Descriptive;
perl -MTie::IxHash -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Tie::IxHash;
perl -MAlgorithm::Combinatorics -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Algorithm::Combinatorics;
perl -MDevel::Size -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Devel::Size;
perl -MSort::Key::Radix -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Sort::Key::Radix;
perl -MSort::Key -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Sort::Key;
perl -MBit::Vector -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext Bit::Vector;
perl -M"feature 'switch'" -e 1 > /dev/null 2>&1 || cpanm -v --notest -l ext feature;
下面是解释为什么这样可以解决问题!!!
What follows is a brief explanation of what the commands above do.
wget -O- http://cpanmin.us fetches the latest version of cpanm and prints it to STDOUT which is then piped to perl - -l ~/perl5 App::cpanminus local::lib. The first - tells perl to expect the program to come in on STDIN, this makes perl run the version of cpanm we just downloaded.perl passes the rest of the arguments to cpanm. The -l ~/perl5 argument tells cpanm where to install Perl modules, and the other two arguments are two modules to install. [App::cpanmins]1 is the package that installs cpanm. local::lib is a helper module that manages the environment variables needed to run modules in local directory.
After those modules are installed we run
eval `perl -I ~/perl5/lib/perl5 -Mlocal::lib`to set the environment variables needed to use the local modules and then
echo 'eval `perl -I ~/perl5/lib/perl5 -Mlocal::lib`' >> ~/.profileto ensure we will be able to use them the next time we log in.
echo 'export MANPATH=$HOME/perl5/man:$MANPATH' >> ~/.profilewill hopefully cause man to find the man pages for your local modules.
这种类似的问题被问的特别多!
There's the way documented in perlfaq8, which is what local::lib is doing for you.
It's also a frequently asked StackOverflow question:
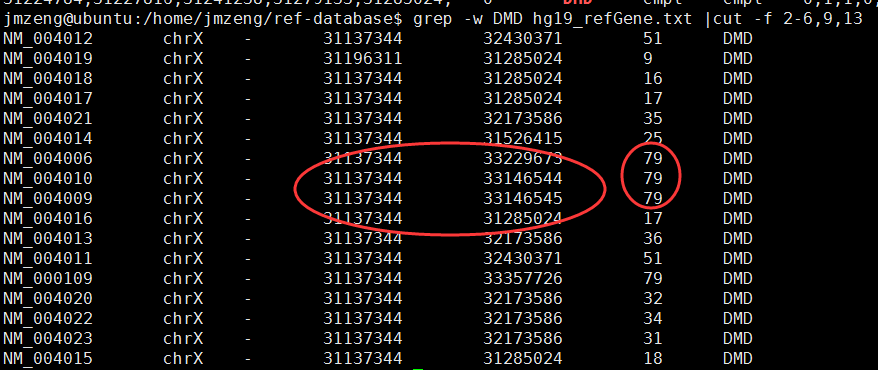
我是受到了SOAPfuse的启发才想到整理各种基因组版本的对应关系,完整版!!!
以后再也不用担心各种基因组版本混乱了,我还特意把所有的下载链接都找到了,可以下载任意版本基因组的基因fasta文件,gtf注释文件等等!!!
GRCh36 (hg18): ENSEMBL release_52.GRCh37 (hg19): ENSEMBL release_59/61/64/68/69/75.GRCh38 (hg38): ENSEMBL release_76/77/78/80/81/82.
Feb 13 2014 00:00 Directory April_14_2003 Apr 06 2006 00:00 Directory BUILD.33 Apr 06 2006 00:00 Directory BUILD.34.1 Apr 06 2006 00:00 Directory BUILD.34.2 Apr 06 2006 00:00 Directory BUILD.34.3 Apr 06 2006 00:00 Directory BUILD.35.1 Aug 03 2009 00:00 Directory BUILD.36.1 Aug 03 2009 00:00 Directory BUILD.36.2 Sep 04 2012 00:00 Directory BUILD.36.3 Jun 30 2011 00:00 Directory BUILD.37.1 Sep 07 2011 00:00 Directory BUILD.37.2 Dec 12 2012 00:00 Directory BUILD.37.3
1. Navigate to http://genome.ucsc.edu/cgi-bin/hgTables2. Select the following options:
clade: Mammal
genome: Human
assembly: Feb. 2009 (GRCh37/hg19)
group: Genes and Gene Predictions
track: UCSC Genes
table: knownGene
region: Select "genome" for the entire genome.
output format: GTF - gene transfer format
output file: enter a file name to save your results to a file, or leave blank to display results in the browser3. Click 'get output'.
for i in $(seq 1 22) X Y M;
do echo $i;
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/chromosomes/chr${i}.fa.gz;## 这里也可以用NCBI的:ftp://ftp.ncbi.nih.gov/genomes/M_musculus/ARCHIVE/MGSCv3_Release3/Assembled_Chromosomes/chr前缀
done
gunzip *.gz
for i in $(seq 1 22) X Y M;
do cat chr${i}.fa >> hg19.fasta;
done
rm -fr chr*.fasta
开发单位:华大,SOAP系列软件套装!
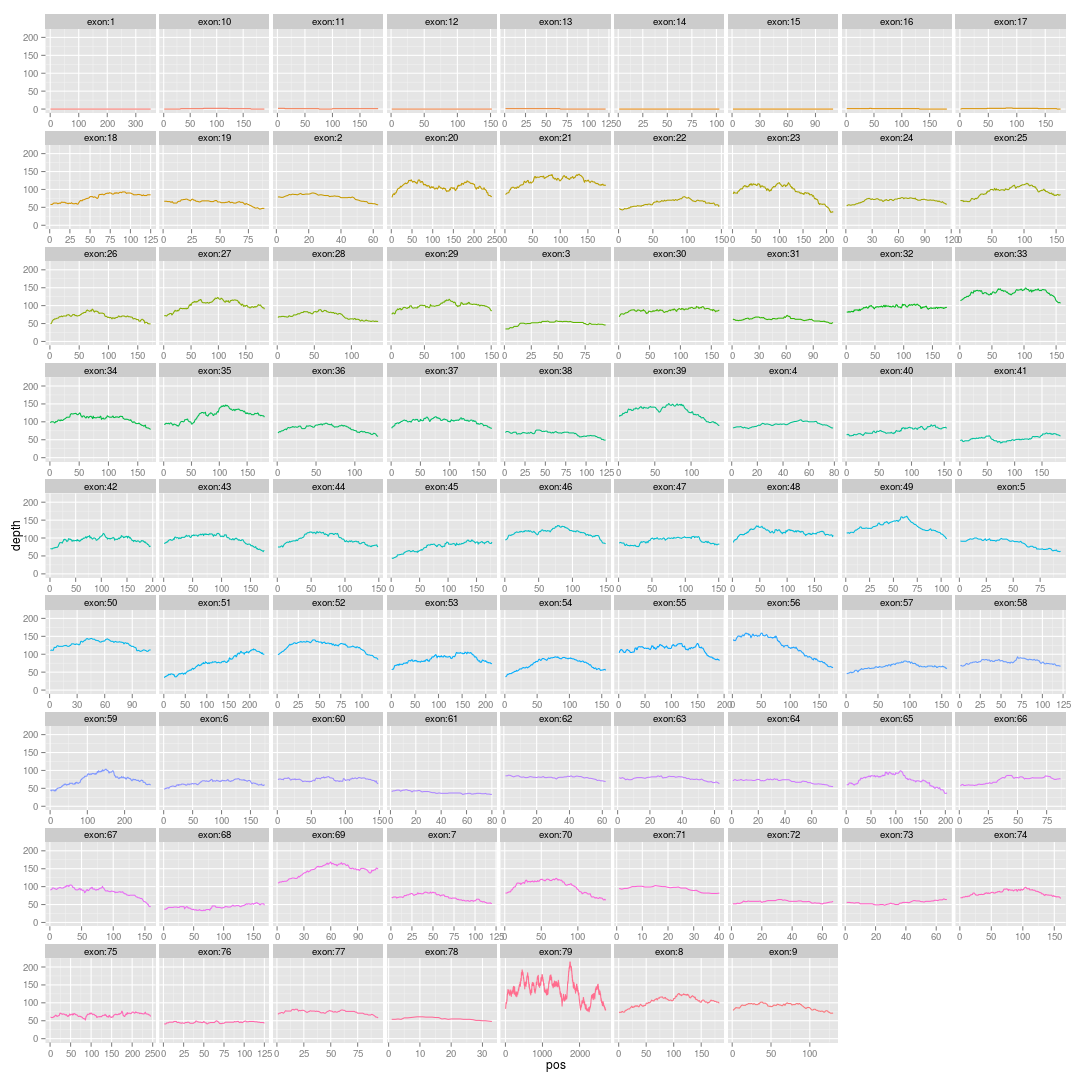
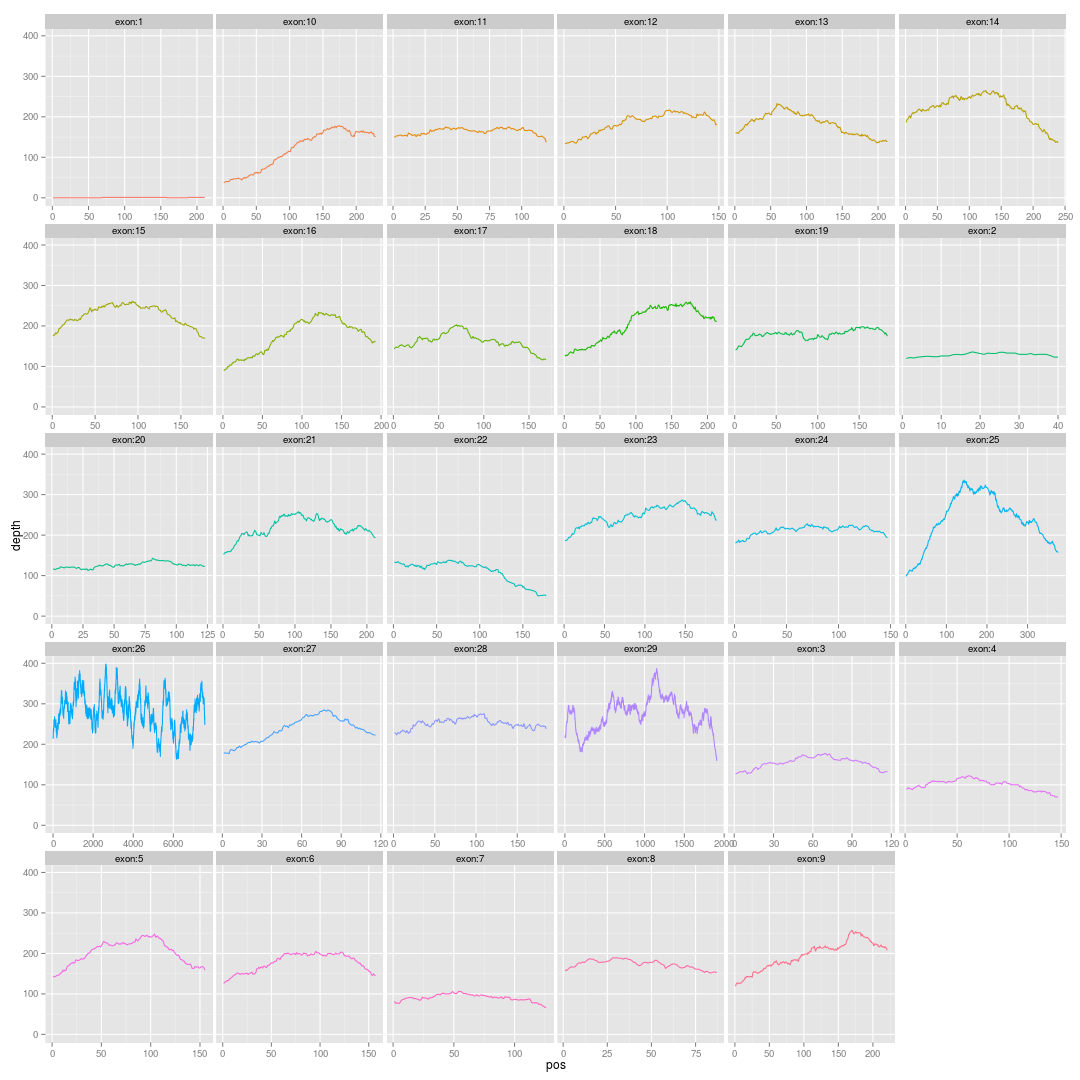
二,输入数据准备
6.5K Jun 15 2009 cytoBand.txt.gz3.0G Oct 12 2012 hg19.fa2.5M Mar 15 10:30 HGNC_Gene_Family_dataset38M Feb 8 2014 Homo_sapiens.GRCh37.75.gtf.gz202 Jan 19 16:07 HumanRef_refseg_symbols_relationship.list
文件下载地址,作者已经给出了!
这一步耗时很长,4~6小时,创造了transcript.fa和gene.fa,然后还对他们建立bwa和soap的index,所以有点慢!
Congratulations!You have constructed SOAPfuse database files successfully.These database files are all stored in directory you supplied:/home/jmzeng/biosoft/SOAPfuse/db_v1.27/They are all generated based on public data files you supplied:whole_genome_fasta_file: /home/jmzeng/biosoft/SOAPfuse/db_v1.27/raw/hg19.fagtf_annotation_file: /home/jmzeng/biosoft/SOAPfuse/db_v1.27/raw/Homo_sapiens.GRCh37.75.gtf.gzChr_Bandregion_file: /home/jmzeng/biosoft/SOAPfuse/db_v1.27/raw/cytoBand.txt.gzHGNC_gene_family_file: /home/jmzeng/biosoft/SOAPfuse/db_v1.27/raw/HGNC_Gene_Family_datasetgtf_segname2refseg_list: /home/jmzeng/biosoft/SOAPfuse/db_v1.27/raw/HumanRef_refseg_symbols_relationship.list
DB_db_dir = /DATABASE_DIR/PG_pg_dir = /TOOL_DIR/source/binPS_ps_dir = /TOOL_DIR/sourcePD_all_out = /out_directory/PA_all_fq_postfix = PostFix
三,运行命令
perl SOAPfuse-RUN.pl -c <config_file> -fd <WHOLE_SEQ-DATA_DIR> -l <sample_list> -o <out_directory> [Options]
运行的非常慢!!!
四,数据结果解读
我已经构建好bioconductor中文社区的雏形,大家可以进去看看!
突然发现我的bioconductor.cn这个域名都快要过期了!
哈哈,才想起一年前的计划到现在还没开始实施,实在不像我的风格,可能是水平到了一定程度吧,很多初级工作不像以前那样事无巨细的把关了。正好,借这个机会找几个朋友帮我一起完成这个bioconductor中文社区计划!
Hi,all. I just started to use github and found this magic way to write a blog.
so I do a simpler test here.
Don't be confused, there is nothing new for bioinformatic here.
And it very easy to use Markdown in wordpress,just download a plugin.
Below is some useful links to help you familar with markdown as soon as possible.
- http://bhttp://mahua.jser.me/log.csdn.net/kaitiren/article/details/38513715
- https://github.com/guodongxiaren/README/blob/master/README.md
- http://wowubuntu.com/markdown/
- http://mahua.jser.me/
Also I want to recommend some useful web-editor for markdown.
- http://mahua.jser.me/
- https://www.madoko.net/
- https://www.zybuluo.com/mdeditor
It's easy to use Markdown, but to get the most out of it, you still need to understand it and keep practising
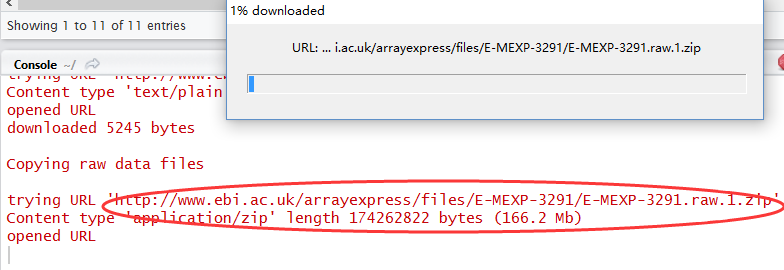
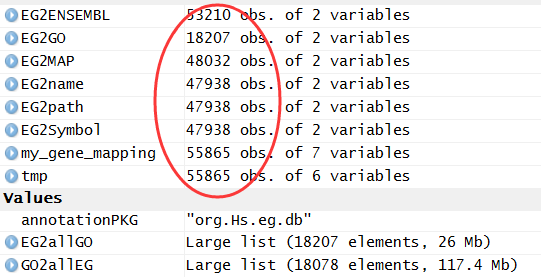
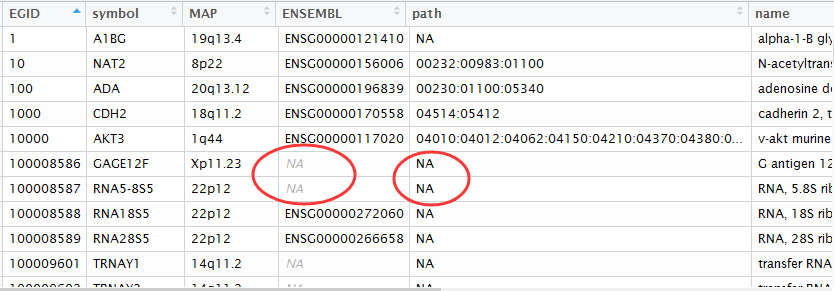
rawset = ArrayExpress("E-MEXP-3291")
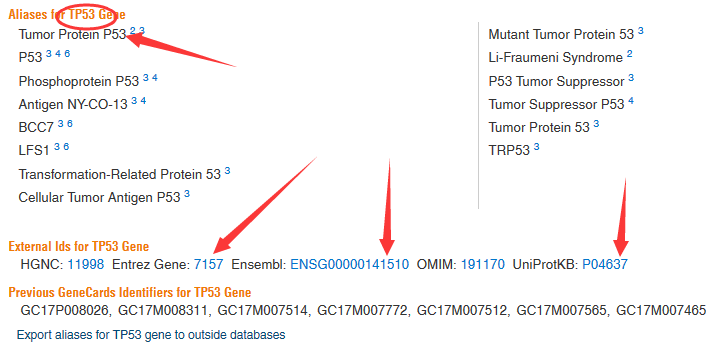
library(org.Hs.eg.db )
This online tool currently uses CrossMap, which supports a limited number of formats (see our online documentation for details of the individual data formats listed below). CrossMap also discards metadata in files, so track definitions, etc, will be lost on conversion.
Important note: CrossMap converts WIG files to BedGraph internally for efficiency, and also outputs them in BedGraph format.
因为大部分生物信息学软件都是linux版本的,所以生物信息学数据分析工作者必备技能就是linux,但是大部分人只是拿他当个中转站,我以前也是,直到接触了大批量的任务,自动化流程,才明白这里面的水太深了,不过无所谓,凭我个人的观点,其实shell的进阶语法真的不必要!
我也不想去搞明白操作符两边是否加空格的区别是什么了。
如果,还有其它功能需要实现,我们可以把脚本写的更负载,纯粹的用shell,需要搜索更多的shell技巧。
[perl]
## perl nohup.pl deep_count.sh 0
## perl nohup.pl deep_count.sh 1
## perl nohup.pl deep_count.sh 2
$i=1;
open FH,$ARGV[0];
while(<FH>){
chomp;
next unless $.%3==$ARGV[1];
$cmd="nohup $_ &";
print "$cmd\n";
system($cmd);
sleep(10800) if $i%5==4;
$i++;
#exit;
}
[/perl]
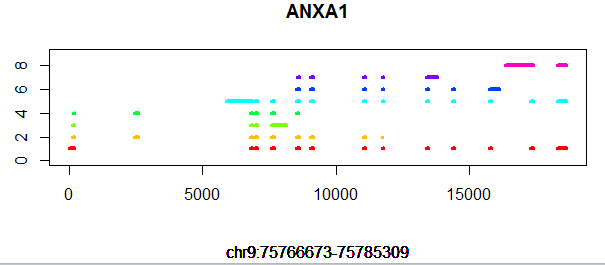
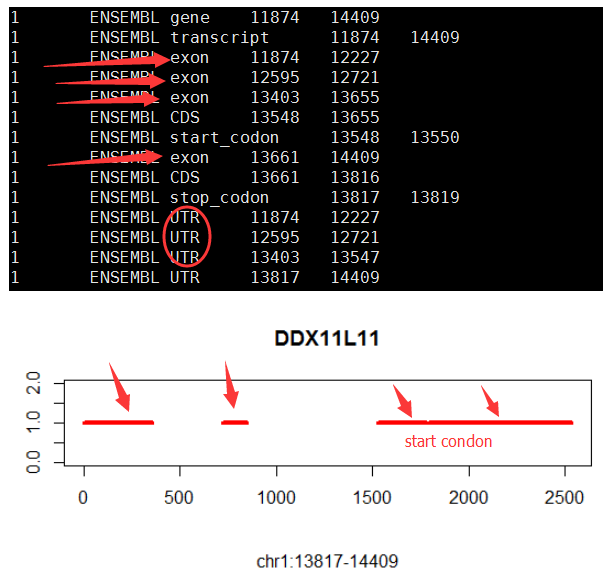
[perl]
suppressMessages(library(ggplot2))
suppressMessages(library(RMySQL))
con <- dbConnect(MySQL(), host="127.0.0.1", port=3306, user="root", password="11111111")
dbSendQuery(con, "USE test")
gene='SOX10'
#gene='DDX11L11'
if (T){
query=paste("select * from hg19_gtf where gene_type='protein_coding' and gene_name=",shQuote(gene),sep="")
structure=dbGetQuery(con,query)
tmp_min=min(c(structure$start,structure$end))
structure$new_start=structure$start-tmp_min
structure$new_end=structure$end-tmp_min
tmp_max=max(c(structure$new_start,structure$new_end))
num_transcripts=nrow(structure[structure$record=='transcript',])
tmp_color=rainbow(num_transcripts)
x=1:tmp_max;y=rep(num_transcripts,length(x))
#x=10000:17000;y=rep(num_transcripts,length(x))
plot(x,y,type = 'n',xlab='',ylab = '',ylim = c(0,num_transcripts+1))
title(main = gene,sub = paste("chr",tmp$chr,":",tmp$start,"-",tmp$end,sep=""))
j=0;
tmp_legend=c()
for (i in 1:nrow(structure)){
tmp=structure[i,]
if(tmp$record == 'transcript'){
j=j+1
tmp_legend=c(tmp_legend,paste("chr",tmp$chr,":",tmp$start,"-",tmp$end,sep=""))
}
if(tmp$record == 'exon') lines(c(tmp$new_start,tmp$new_end),c(j,j),col=tmp_color[j],lwd=4)
}
# legend('topleft',legend=tmp_legend,lty=1,lwd = 4,col = tmp_color);
}
[/perl]
刚开始接触生物信息学的时候我也很纠结什么fastq,fastq,sam,bam,vcf,maf,gtf,bed,psl等等,甚至还有过时了的NCBI,ENSEMBL格式,如果是我刚开始 学的时候,我倒是很愿意把他们全部搞透彻,写详细的说明书,但是现在成长了,这些东西感觉很low了,正好我看到了一篇帖子讲数据格式的收集大全,分享给大家,希望初学者能多花点时间好好钻研!
https://www.biostars.org/p/55351/
现有的基因芯片种类不要太多了!
gpl organism bioc_package1 GPL32 Mus musculus mgu74a2 GPL33 Mus musculus mgu74b3 GPL34 Mus musculus mgu74c6 GPL74 Homo sapiens hcg1107 GPL75 Mus musculus mu11ksuba8 GPL76 Mus musculus mu11ksubb9 GPL77 Mus musculus mu19ksuba10 GPL78 Mus musculus mu19ksubb11 GPL79 Mus musculus mu19ksubc12 GPL80 Homo sapiens hu680013 GPL81 Mus musculus mgu74av214 GPL82 Mus musculus mgu74bv215 GPL83 Mus musculus mgu74cv216 GPL85 Rattus norvegicus rgu34a17 GPL86 Rattus norvegicus rgu34b18 GPL87 Rattus norvegicus rgu34c19 GPL88 Rattus norvegicus rnu3420 GPL89 Rattus norvegicus rtu3422 GPL91 Homo sapiens hgu95av223 GPL92 Homo sapiens hgu95b24 GPL93 Homo sapiens hgu95c25 GPL94 Homo sapiens hgu95d26 GPL95 Homo sapiens hgu95e27 GPL96 Homo sapiens hgu133a28 GPL97 Homo sapiens hgu133b29 GPL98 Homo sapiens hu35ksuba30 GPL99 Homo sapiens hu35ksubb31 GPL100 Homo sapiens hu35ksubc32 GPL101 Homo sapiens hu35ksubd36 GPL201 Homo sapiens hgfocus37 GPL339 Mus musculus moe430a38 GPL340 Mus musculus mouse430239 GPL341 Rattus norvegicus rae230a40 GPL342 Rattus norvegicus rae230b41 GPL570 Homo sapiens hgu133plus242 GPL571 Homo sapiens hgu133a243 GPL886 Homo sapiens hgug4111a44 GPL887 Homo sapiens hgug4110b45 GPL1261 Mus musculus mouse430a249 GPL1352 Homo sapiens u133x3p50 GPL1355 Rattus norvegicus rat230251 GPL1708 Homo sapiens hgug4112a54 GPL2891 Homo sapiens h20kcod55 GPL2898 Rattus norvegicus adme16cod60 GPL3921 Homo sapiens hthgu133a63 GPL4191 Homo sapiens h10kcod64 GPL5689 Homo sapiens hgug4100a65 GPL6097 Homo sapiens illuminaHumanv166 GPL6102 Homo sapiens illuminaHumanv267 GPL6244 Homo sapiens hugene10sttranscriptcluster68 GPL6947 Homo sapiens illuminaHumanv369 GPL8300 Homo sapiens hgu95av270 GPL8490 Homo sapiens IlluminaHumanMethylation27k71 GPL10558 Homo sapiens illuminaHumanv472 GPL11532 Homo sapiens hugene11sttranscriptcluster73 GPL13497 Homo sapiens HsAgilentDesign02665274 GPL13534 Homo sapiens IlluminaHumanMethylation450k75 GPL13667 Homo sapiens hgu21976 GPL15380 Homo sapiens GGHumanMethCancerPanelv177 GPL15396 Homo sapiens hthgu133b78 GPL17897 Homo sapiens hthgu133a
gpl_info=read.csv("GPL_info.csv",stringsAsFactors = F)### first download all of the annotation packages from bioconductorfor (i in 1:nrow(gpl_info)){print(i)platform=gpl_info[i,4]platform=gsub('^ ',"",platform) ##主要是因为我处理包的字符串前面有空格#platformDB='hgu95av2.db'platformDB=paste(platform,".db",sep="")if( platformDB %in% rownames(installed.packages()) == FALSE) {BiocInstaller::biocLite(platformDB)#source("http://bioconductor.org/biocLite.R");#biocLite(platformDB )}}
下载完了所有的包, 就可以进行批量导出芯片探针与gene的对应关系!
for (i in 1:nrow(gpl_info)){print(i)platform=gpl_info[i,4]platform=gsub('^ ',"",platform)#platformDB='hgu95av2.db'platformDB=paste(platform,".db",sep="")if( platformDB %in% rownames(installed.packages()) != FALSE) {library(platformDB,character.only = T)#tmp=paste('head(mappedkeys(',platform,'ENTREZID))',sep='')#eval(parse(text = tmp))###重点在这里,把字符串当做命令运行all_probe=eval(parse(text = paste('mappedkeys(',platform,'ENTREZID)',sep='')))EGID <- as.numeric(lookUp(all_probe, platformDB, "ENTREZID"))##自己把内容写出来即可}}