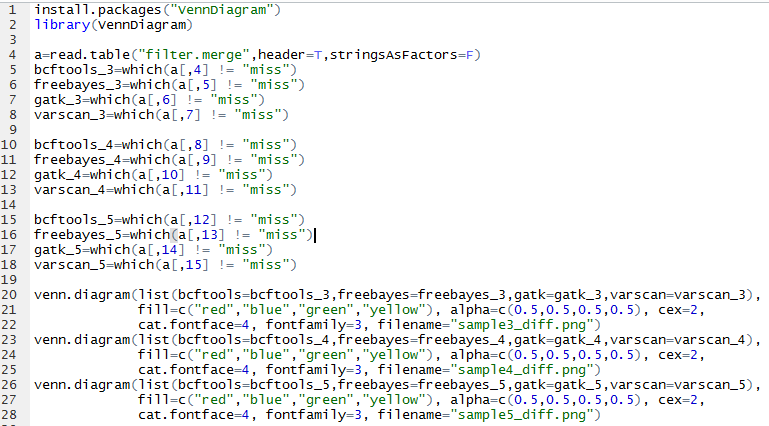
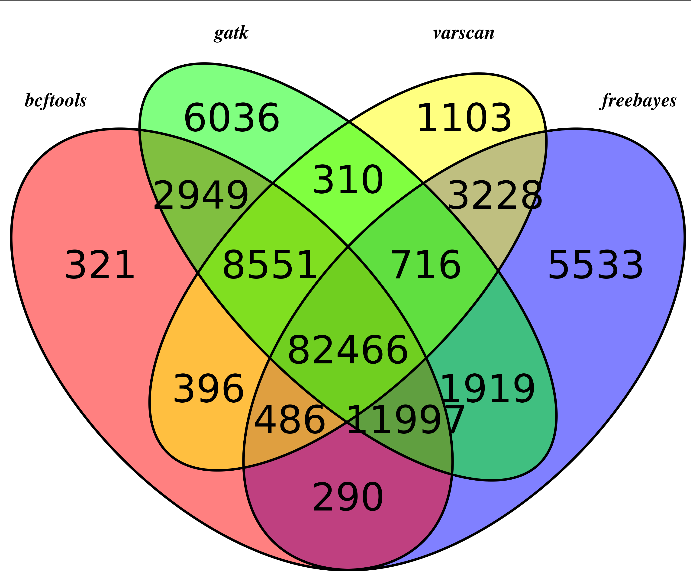
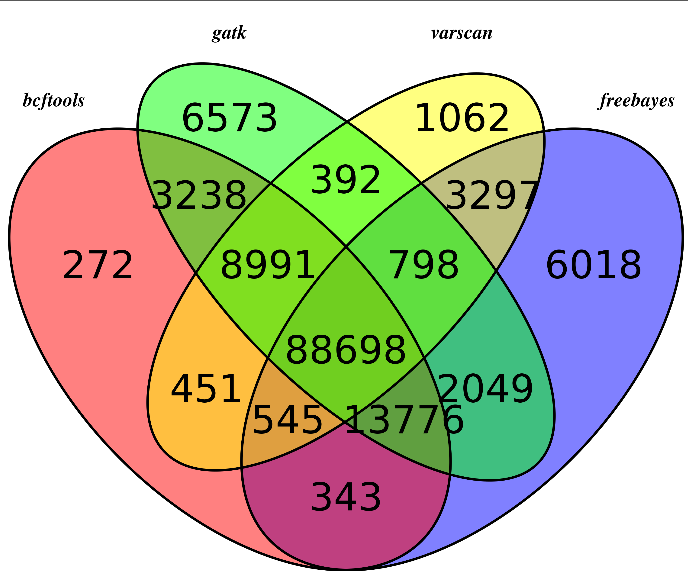
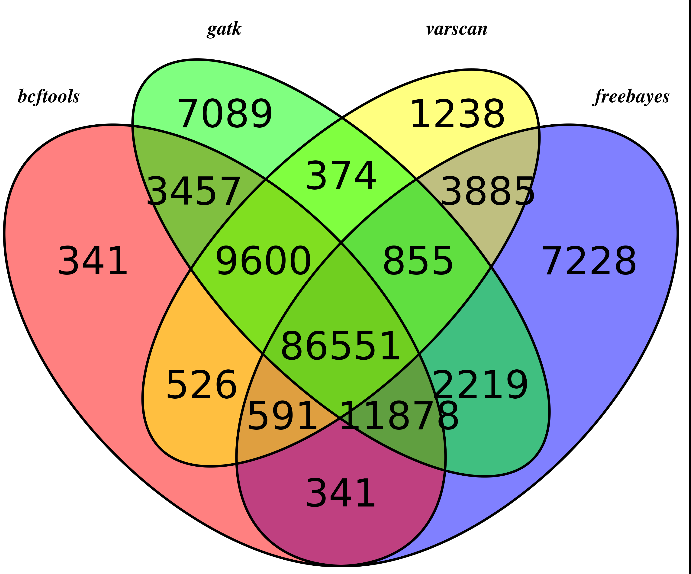
无意中看到这篇文章,感觉挺有趣的!
我不知道国外的教育是咋样的,但是文中描述的中国教育的试题都是千真万确的,我也经历过。
我没看过有钱人家的小孩,不知道那些花了几百万买个几平米的学区房的那些人的小孩是不是也要经受这样的教育摧残?
我还有个问题,我是如何出淤泥而不染的呢?
随便瞎扯,大家自己看这篇文章吧!
可怕的中国教育,聪明伶俐进去呆若木鸡出来
作者:葛小琴
来源:杂文月刊(文摘版)
侄子在读高二,考了一道历史题:成吉思汗的继承人窝阔台,公元哪一年死?最远打到哪里?答不出来,我帮他查找资料,所以到现在我都记得,是打到现在的匈牙利附近。
在一次偶然的机会,我发现美国世界史这道题目不是这样考的。它的题目是这样的:成吉思汗的继承人窝阔台,当初如果没有死,欧洲会发生什么变化?试从经济、政治、社会三方面分析。
有个学生是这样回答的:这位蒙古领导人如果当初没有死,那么可怕的黑死病,就不会被带到欧洲去,后来才知道那个东西是老鼠身上的跳蚤引起的鼠疫。但是六 百多年前,黑死病在欧洲猖獗的时候,谁晓得这个叫做鼠疫?如果没有黑死病,神父跟修女就不会死亡。神父跟修女如果没有死亡,就不会怀疑上帝的存在。如果没 有怀疑上帝的存在,就不会有意大利弗罗伦斯的文艺复兴?
如果没有文艺复兴,西班牙、南欧就不会强大,西班牙无敌舰队就不可能建立。如果西班牙、意大利不够强大,盎格鲁—撒克逊会提早200年强大,日耳曼会控制中欧,奥匈帝国就不可能存在。
教师一看“棒,分析得好。”但他们没有分数,只有等级A。其实这种题目老师是没有标准答案的,可是大家都要思考。
不久前,我去了趟日本,日本总是和我们在历史问题上产生纠葛,所以我在日本很注意高中生的教科书。
他们的教师给高中生布置了这样一道题:日本跟中国100年打一次仗,19世纪打了日清战争(即甲午战争),20世纪打了一场日中战争(即抗日战 争),21世纪如果日本跟中国开火,你认为大概是什么时候?可能的远因和近因在哪里?如果日本赢了,是赢在什么地方?输了是输在什么条件上?分析之。
其中有个高中生是这样分析的:我们跟中国很可能在台湾回到中国以后,有一场激战。台湾如果回到中国,中国会把基隆与高雄封锁,台湾海峡就会变成中国的内 海,我们的油轮就统统走右边,走基隆和高雄的右边。这样,会增加日本的运油成本。我们的石油从波斯湾出来跨过印度洋,穿过马六甲海峡,上中国南海,跨台湾 海峡进东海到日本海,这是石油生命线,中国政府如果把台湾海峡封锁起来,我们的货轮一定要从那里经过,我们的主力舰和驱逐舰就会出动,中国海军一看到日本 出兵,马上就会上场,就开打。
按照判断,公元2015年至2020年之间,这场战争可能爆发。所以,我们现在就要做对华抗战的准备。
我看其他学生的判断,也都是中国跟日本的摩擦会从东海从台湾海峡开始,时间判断是2015年至2020年之间。
这种题目和答案都太可怕了。
撇开政治因素来看这道题,我们的历史教育就很有问题。翻开我们的教科书,题目是这样出的:甲午战争是哪一年爆发的?签订的叫什么条约?割让多少土地?赔 偿多少银两?每个学生都努力做答案。结果我们一天到晚研究什么时候割让辽东半岛,什么时候丢了台湾、澎湖、赔偿二万银两,1894年爆发甲午战争、 1895年签订马关条约,背得滚瓜烂熟,都是一大堆枯燥无味的数字。
那又怎么样,反正都赔了嘛,银两都给了嘛,最主要的是将来可能会怎样。
人家是在培养能力,而我们是在灌输知识,这是值得深思的部分!
看外面的教育,再看我们的教育?
老妈去参加我侄子的家长会,带回了两套侄子的考试试卷,我很好奇,拿过来看了现在小学生的试卷后,我震惊了!这是什么狗屁教育?这样的教育有希望吗?下面给大家详细说说我看到了什么。
侄子在本市某著名小学读书,有这么几道题。
一个春天的夜晚,一个久别家乡的人,望着皎洁的月光不禁思念起了故乡,于是吟起了一首诗:( ),( )。
我看到侄子答的是:举头望明月,低头思故乡。但后面是一把大大的×,我就奇怪了,我也是想到的这两句。好奇地问侄子,这个不对??那答案是什么?侄子说 标准答案是:春风又绿江南岸,明月何时照我还。哎,这就奇怪了,因为是个春天的夜晚,就要是这句有春风的???要这个思念故乡的人不是江南的,是不可能说 出春风又绿江南岸这句话的!!!举头望明月,低头思故乡应该更准确。再扯远点,思念故乡,一千个人可以吟一千句不一样的诗,这个也可以有标准答案的么?
接下来是默写,题目是:我们学过《桂林山水》一文,请将下面句子默写下来。然后就是整段的要默写,这有什么用?死记硬背别人的文字有什么用?
还有个题目,《匆匆》这篇课文,是现代着名作家朱自清先生写的,同学们都很喜欢这篇散文,你能把自己最喜欢,印象最深刻的一句写下来吗?我侄子写的是: 我的日子滴在时间的流里,没有声音,也没有影子。后面一把好大的×。标准答案竟然是:但是,聪明的你告诉我,我们的日子为什么一去不复返呢?这就更奇怪 了,一篇文章,你可以喜欢这句,我可以喜欢那句,难道最喜欢的一句话也要统一么?为什么“我的日子滴在时间的流里,没有声音,也没有影子。”这句不能喜 欢?就一定要喜欢“但是,聪明的你告诉我,我们的日子为什么一去不复返呢?”这句?我觉得这个题目应该是“你能把老师最喜欢,印象最深刻的一句写下来 吗?”才对!
再看别的试卷,更莫名其妙了,比如请说出阿拉伯数字的来历,是哪个国家创造的?侄子不知道,问我,我也不知道。我只好去搜一下,才知道是古印度人发明的。莫非我吃块猪肉,还一定得知道它是哪个养猪场养出来的?
最后有个题目让我彻底崩溃了:请用一句话说明π的含义。侄子回答π的含义是圆周率。竟然打的是×,这就奇怪了,正好我老婆大学说读的是理科,我马上问 她,π是什么意思,她说圆周率啊。两个人狂汗,问了侄子半天,标准答案大概是,π是一个在数学及物理学领域普遍存在的数学常数……
如果你也觉得这种教育很无耻,就请转发吧,让更多的人来参与呼吁改变,为了孩子为了国家的未来 …… 别让孩子聪明伶俐地进去呆若木鸡地出来!