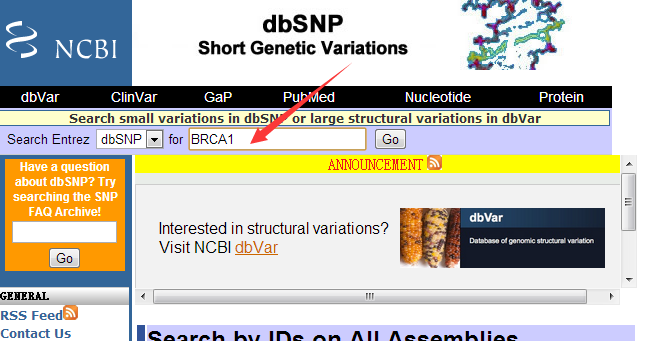
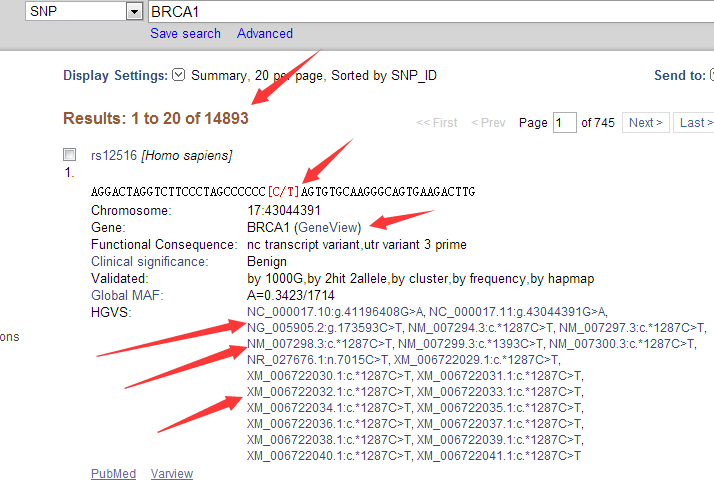
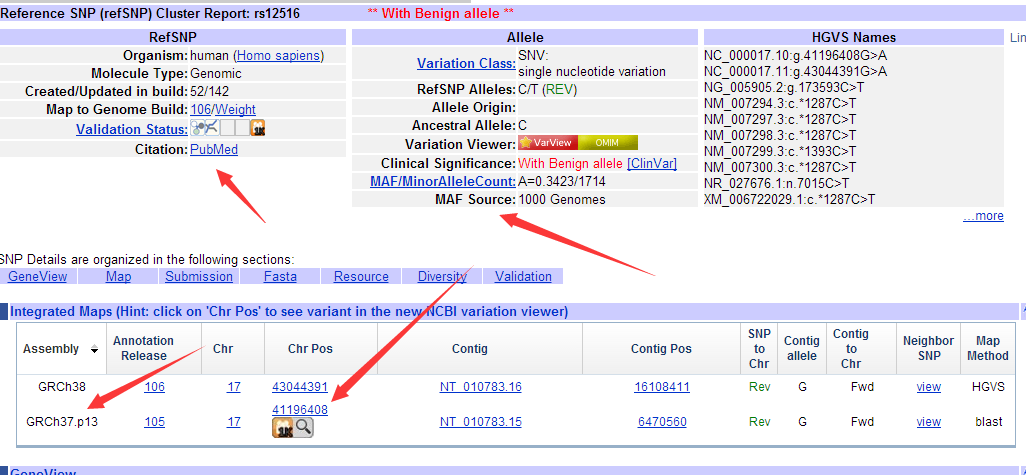
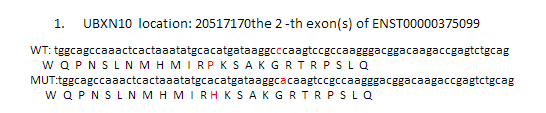至于如何安装该软件,请见上一个教程
一.首先把snp-calling步骤的VCF文件转为annovar软件要求的格式
convert2annovar.pl -format vcf4 12.vcf >12.annovar
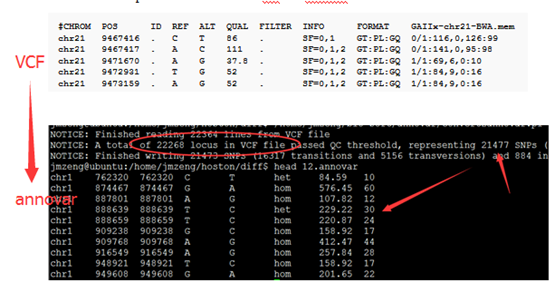
二.进行注释
命令行参数比较多,还是用脚本来运行
# define path
infolder=/home/jmzeng/hoston/diff
outfolder=$infolder
annovardb=/home/jmzeng/bio-soft/annovar/humandb
# start annotating
/home/jmzeng/bio-soft/annovar/annotate_variation.pl \
--buildver hg19 \
--geneanno \
--outfile ${outfolder}/12.anno \
${infolder}/12.annovar \
${annovardb}
三.输出结果解读
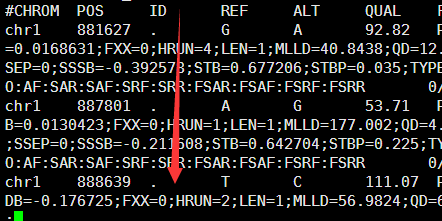
2.6M Apr 14 22:32 12.anno.exonic_variant_function
1.9K Apr 14 22:32 12.anno.log
1.3M Apr 14 22:32 12.anno.variant_function
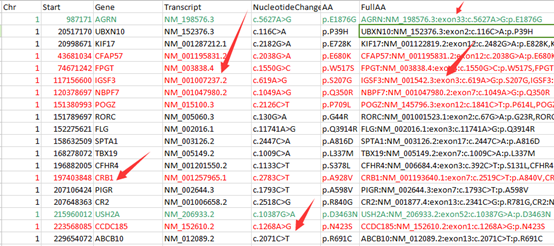
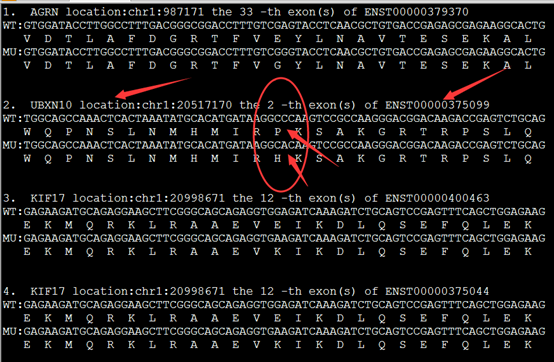
重点是后缀为exonic_variant_function,这个文件对每一个vcf的突变都进行了注释。
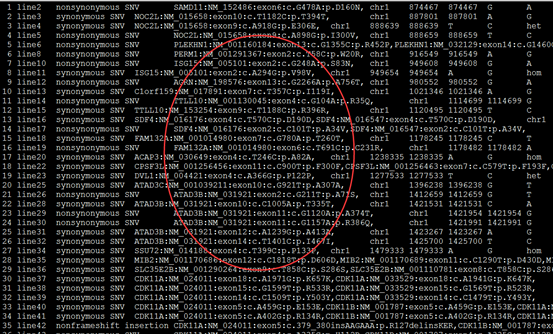
这个结果就可以用来解析了,可以根据实验设计来找到自己感兴趣的突变。
第5.6列是染色体及pos坐标
第4列信息非常复杂,是突变的注释
第12列是测序深度,一般要大于20
我这里是先把注释文件转换成以下格式
location:chr1:874467 SAMD11:NM_152486:exon6:c.G478A:p.D160N
location:chr1:888639 NOC2L:NM_015658:exon9:c.A918G:p.E306E
location:chr1:888659 NOC2L:NM_015658:exon9:c.A898G:p.I300V
location:chr1:916549 PERM1:NM_001291367:exon2:c.T58C:p.W20R
location:chr1:949608 ISG15:NM_005101:exon2:c.G248A:p.S83N
location:chr1:980552 AGRN:NM_198576:exon13:c.G2266A:p.A756T
location:chr1:1114699 TTLL10:NM_001130045:exon4:c.G104A:p.R35Q
location:chr1:1158631 SDF4:NM_016176:exon4:c.T570C:p.D190D
location:chr1:1158631 SDF4:NM_016547:exon4:c.T570C:p.D190D
location:chr1:1164073 SDF4:NM_016176:exon2:c.C101T:p.A34V
然后比较两个文件,取不同的突变来格式化输出。