下载地址我就不贴了,随便谷歌一下即可!
Genome Reference Consortium Human ---》 GRCh3
Feb. 2009 (hg19, GRCh37)这个是重点
Mar 2006 assembly = hg18 = NCBI36.
May 2004 assembly = hg17 = NCBI35.
July 2003 assembly = hg16 = NCBI34
以前的老版本就不用看啦,现在其实都已经有hg38出来啦,GRCh38 (NCBI) and hg38(UCSC)
参考:http://age.wang.blog.163.com/blog/static/119252448201092284725460/
http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/
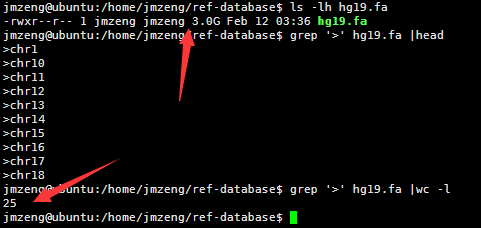
人的hg19基因组是3G的大小,因为一个英文字符是一个字节,所以也是30亿bp的碱基。
包括22条常染色体和X,Y性染色体及M线粒体染色体。
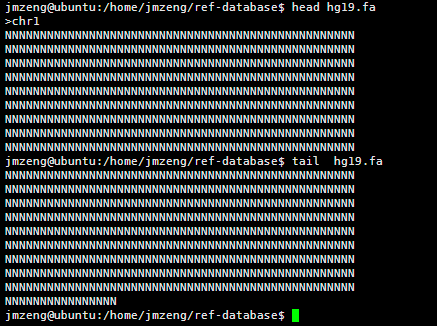
查看该文件可以看到,里面有很多的N,这是基因组里面未知的序列,用N占位,但是觉得部分都是A.T.C.G这样的字符,大小写都有,分别代表不同的意思。
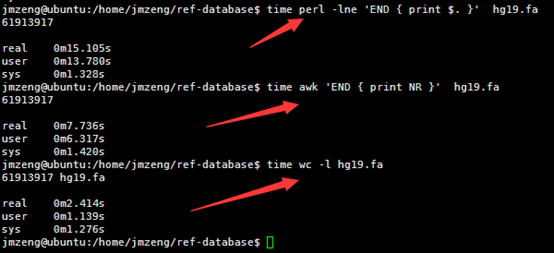
然后我用linux的命令统计了一下里面这个文件的行数,
perl -lne 'END { print $. }' hg19.fa
awk 'END { print NR }' hg19.fa
wc -l hg19.fa
然后我写了一个脚本统计每条染色体的长度,42秒钟完成任务!
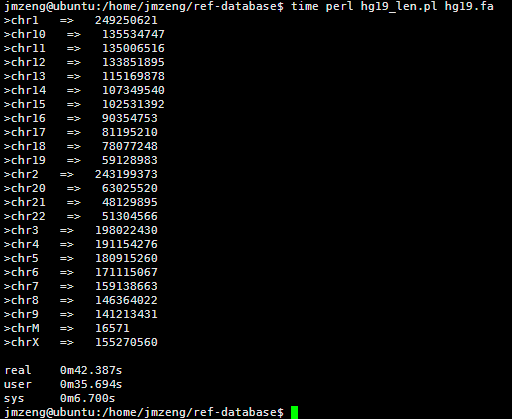
看来这个服务器的性能还是蛮强大的,读取文件非常快!
[perl]
while(<>){
chomp;
if (/>/){
if (exists $hash_chr{$key} ){
$len = length $hash_chr{$key};
print "$key => $len\n";
}
undef %hash_chr;
$key=$_;
}
else {
$hash_chr{$key}.=$_;
}
}
[/perl]
然后我用seed统计了一下hg19的词频(我不知道生物信息学里面的专业描述词语是什么)
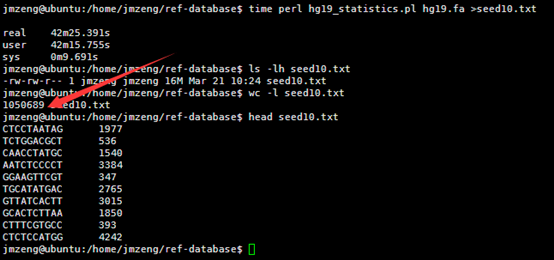
我的程序耗费了42分钟才跑完,感觉我写的程序应该是没有问题的,让我吃惊的是总共竟然只有105万条独特的10bp短序列。然后我算了一下4的10次方,(⊙o⊙)…悲剧,原来只有1048576,之所以出现这种情况,是因为里面有N这个字符串,不仅仅是A.T.C.G四个字符。我用grep -v N seed10.txt |wc -l命令再次统计了一下,发现居然就是1048576,也就是说,任意A.T.C.G四个字符组成的10bp字符串短序列在人的基因组里面都可以找到!!!
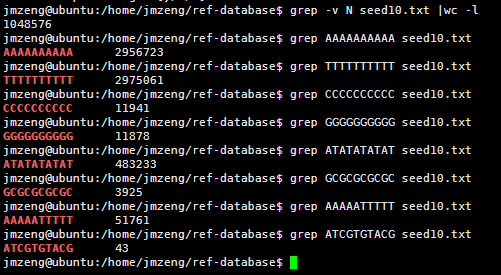
然后我测试了一下,还是真是这样的,真是一个蛮有意思的现象。虽然我无法解释为什么,但是根据这个结果我们可以得知连续的A或者T在人类基因组里面高频出现,而连续的G或者C却很少!
如果我们储存这个10bp字符串的同时,也储存着它们在基因组的位置,那么就可以根据这个seed来进行比对,这就是blast的原理之一!