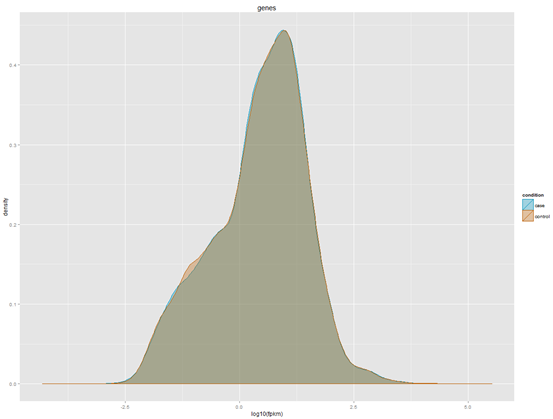
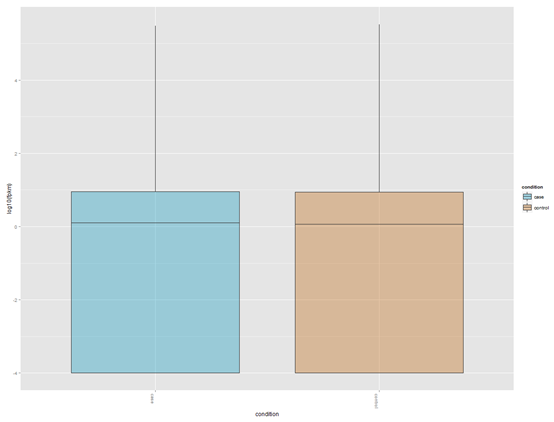
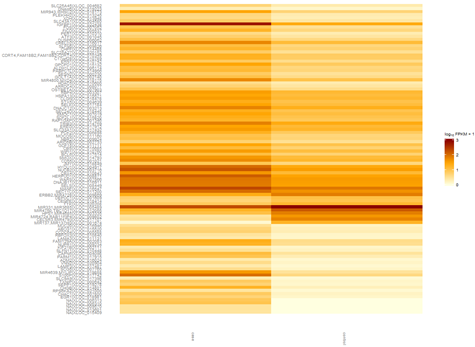
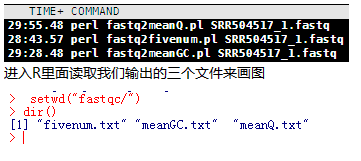
YGC是我非常喜欢的一个博客,但是之前都是英文,所以我没怎么看,余老师还在R领域很有建树,个人发表了5个R在生信方面的包,我后面有空会一个个试用学习。
本文转自http://ygc.name/2014/08/27/insertion-size/
里面详细解释了NGS测序的几个基本概念,也是我之前一直弄混淆的概念,包括插入片段、单端测序,双端测序,配对测序,contig,scaffold等等
在进行测序的时候,需要将DNA打断,构建library,这些fragment需要接上adaptor,好进行扩增,illumina的测序,可以有single end和paired end两种,分别从一端和两端进行测序。
fragment ======================================== fragment + adaptors ~~~========================================~~~ SE read ---------> PE reads R1---------> <---------R2 unknown gap ....................
insertion并不是指R1和R2之间的unknown gap,早在NGS之前,当我们在使用ecoli构建载体的时候,这个概念就已经形成,它是adaptors之间的序列。而unknown gap则称之为inner mate:
PE reads R1---------> <---------R2 fragment ~~~========================================~~~ insert ======================================== inner mate ....................
显然我们不希望看到大量的unknown gap,所以要制造短的fragment,而且技术不断发展,测序长度也越来越长,于是可以测通fragment:
fragment ~~~========================================~~~ insert ======================================== R1 -------------------------> R2 <----------------------- overlap :::::::::: stitched SE read --------------------------------------->
这样R1和R2就有overlap,合并一致序列,就可以得到完整的fragment,使用短的fragment,也就是insertion size比较小的library,测序的结果coverage比较大,因为我们可以测通fragment.
虽然adaptor不会被测序,但如果fragment太短,被读通了,则另一端的adaptor就会被测到。
tiny fragment ~~~~========================~~~~ insert ======================== R1 --------------------------> R2 <-------------------------- read-through !!! !!!
如果MiSeq设置正确的话,读通的adaptor是会被切除了,这样就会获得长度不一致的short reads,也可以使用N来替换adaptor序列,这样长度一样,但会在5' end看到很多N。如果没设置好,reads里含有adaptor序列,那么必须要通过软件去除,否则后续的分析都会有问题。
所以insertion size小有个好处,测序的genome coverage高,但是在进行de novo assembly的时候,有一个问题,如果基因组含有比read length还要长的重复元件时,就无法拼接,所以得到的是很多的contigs,它们之间的gap要长于insertion size且无法确定。这个问题是相当普遍的,即使是相对简单的ecoli基因组,也有一定数量的重复元件。
这个问题需要使用大的insertion size进行paired end测序来解决。
fragment + adaptors ~~~========================================~~~ PE reads R1---------> <---------R2 unknown gap ....................
在这种insertion size比较大的情况下,我们可以估计R1和R2之间的距离,只要有一个片段能够被mapped到unique position的话,那么另一个片段的大致位置就可以确定。所以为了达到好的拼接效果,长fragment的library也是必须的,它有可能给出 contigs间的相对位置。
所以理想的情况是使用multiple insert libraries,short-insert library可以保证获得足够的coverage,它可以告诉你contigs之间的序列,但信息是local的,它没办法告诉你怎么拼;而long- insert libary则可以告诉你一些相对global的信息。

在上面这个测试的数据中,加了long-insert libary虽然在coverage上没多少变化,但N50和最大的contig都显著提高,4.5Mb已经覆盖了~98%的ecoli基因组。
- See more at: http://ygc.name/2014/08/27/insertion-size/#sthash.fKmWKoaf.dpuf