很多时候,我们都没办法很快判断seurat默认聚类分群后的每个亚群的生物学命名,会短暂的把大家先归纳为一个大类,比如肿瘤单细胞数据第一次分群通用规则,按照 : Continue reading
六
25
很多时候,我们都没办法很快判断seurat默认聚类分群后的每个亚群的生物学命名,会短暂的把大家先归纳为一个大类,比如肿瘤单细胞数据第一次分群通用规则,按照 : Continue reading
有人提问,他自己做单细胞的gsva, 细胞通讯,转录因子,拟时序, inferCNV这些分析,发现特别的消耗计算资源,因为项目很多,每个细胞亚群都是过万的细胞。希望可以这些单细胞亚群进行抽样,使得其细胞数量一致。 Continue reading
我们生信技能树已经有多位大神发表了自己的网页工具,其中基于R语言的shiny框架是比较适合初学者的,而且手把手的教程不少: Continue reading
看到单细胞转录组测序数据的文献:《Single-cell sequencing links multiregional immune landscapes and tissue-resident T cells in ccRCC to Continue reading
客户寄送了80个10X单细胞转录组样品的fastq数据给我,本来是想着恰好五一把cellranger流程挂起来,我们也是在单细胞天地公众号详细介绍了cellranger全部使用细节及流程,大家可以自行前往学习,如下: Continue reading
我最近看到了一个综述,在讨论 TNFSF15 基因 ,亦称TL1A或VEGI(血管内皮细胞生长抑制因子),为肿瘤坏死因子超家族成员之一。 Continue reading
年前立下的flag,说要把明码标价专栏扩充到100个项目: Continue reading
如果你也是hg19和hg38傻傻的分不清,可以先看看我五年前的博客介绍: Continue reading
眼尖的小伙伴已经看到了我们的b站免费视频课程《临床生存分析》啦,而且开始发邮件给我申请课程配套代码和数据!
Continue reading
前面的明码标价服务以数据分析为主: Continue reading
最近学员群又有人问到了 Agilent-012391 Whole Human Genome Oligo Microarray G4112A 这样的芯片数据,我让学生打包数据成为rdata发给我,我检查了一下,发现里面的基因ID其实是有问题的,如下所示: Continue reading
最近有粉丝在我们《生信技能树》公众号后台提出来了一个很有意思的问题, 他做的是2X3X3=18个样品的转录组测序,做完了各种各样的组合的差异分析,也做了WGCNA,想多加一个花样,就是最近看到的蛋白编码基因和非编码基因的表达量相关性探索。 Continue reading
最近咱们《生信技能树》的VIP答疑群,有这样的提问: Continue reading
最近刷了刷植物领域单细胞文献,有一个蛮早期的拟南芥根部单细胞研究:《High-Throughput Single-Cell Transcriptome Profiling of Plant Cell Types》对拟时序分析描述的很清楚,适合做科普! Continue reading
众所周知10x单细胞会给出3个文件,我在单细胞数据分析的基础10讲写的很清楚: Continue reading
猪马牛羊狗等动物的科学研究貌似并不多,最近接到《生信技能树》公众号后台粉丝提问,跟着我们的转录组课程没办法完成自己的数据分析,因为物种不一样。 Continue reading
数据挖掘的本质是把基因的数量搞小,而数据挖掘课题的开启核心就是分组,你可以根据容易基因的高低表达量或者甲基化与否,突变与 Continue reading
在生信技能树的教程:《你确定你的差异基因找对了吗?》, 提到过,必须要对你的转录水平的全局表达矩阵做好质量控制,最好是看到标准3张图:
- 左边的热图,说明我们实验的两个分组,normal和npc的很多基因表达量是有明显差异的
- 中间的PCA图,说明我们的normal和npc两个分组非常明显的差异
- 右边的层次聚类也是如此,说明我们的normal和npc两个分组非常明显的差异
如果分组在3张图里面体现不出来,我们是警告大家如果强行进行后续差异分析是有风险的。但并不意味着这样就没办法进行后续分析,我在教程:PCA都分不开的两个组强行找差异是为何提到过无数的这样的例子!
而且呢,本来你自己实验设计好分成两组就不一定是有全局差异,比如文章:ATAC-seq reveals alterations in open chromatin in pancreatic islets from subjects with type 2 diabetes. Sci Rep 2019 May 23;9(1):7785. PMID: 31123324,其配套数据集在:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE129383,实验材料是Human islets of 9 non-diabetic donors and 6 donors diagnosed with T2D,也就是说是15个样品的ATAC-seq数据。
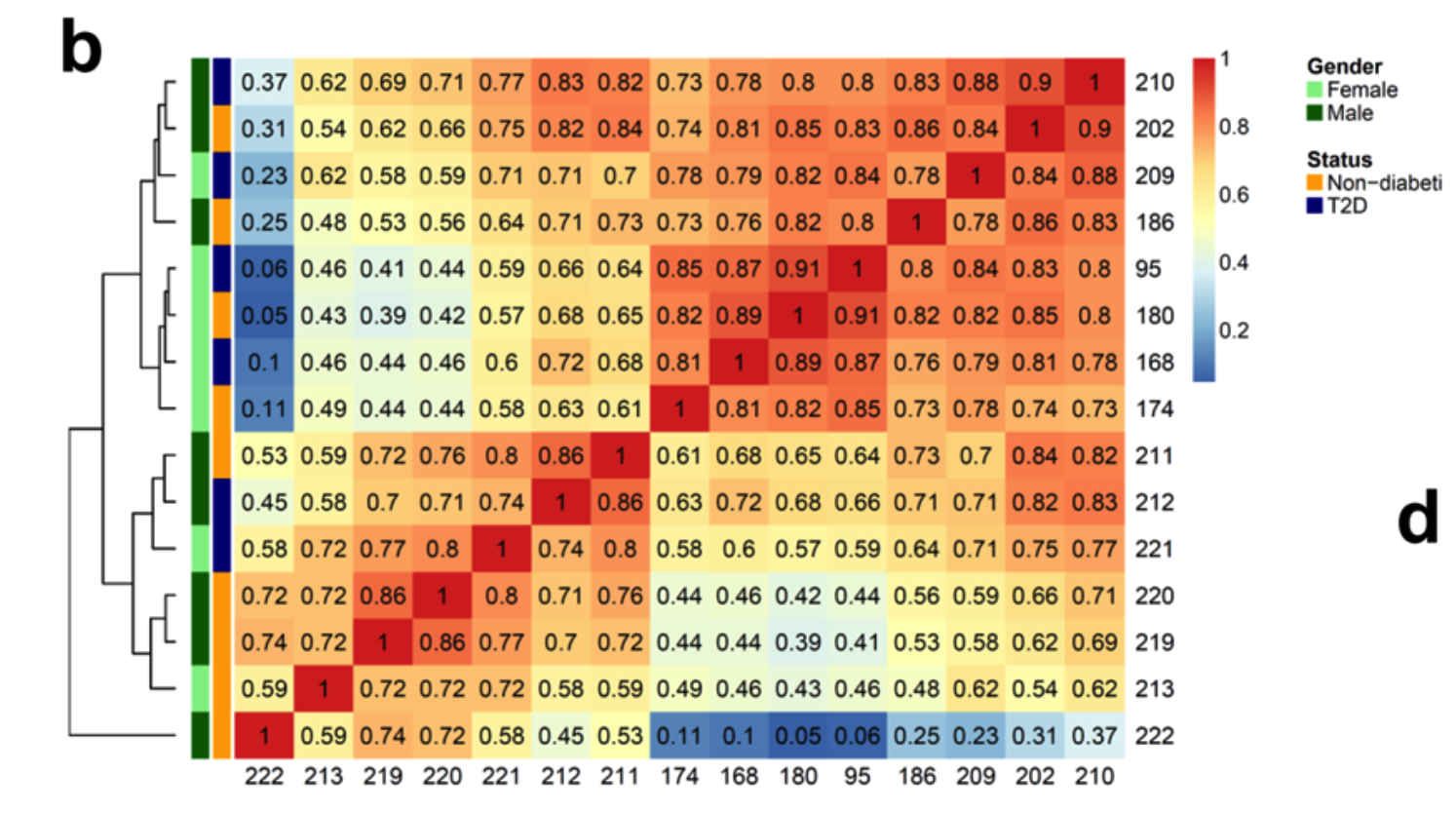
首先看15个样品的ATAC-seq数据全局差异,如下所示 :
如果采用我们转录组授课提到的3张图标准,可以看到这个组内差异和组间差异其实是混淆的。糖尿病患者和正常人勉强还是可以区分开来,但是混杂因素有点多,而且可以肯定这个混杂因素并不是性别,有可能是纳入的病人的年龄或者其它基础疾病,总之呢仅仅是一个糖尿病无法彻底把样品分成两组!
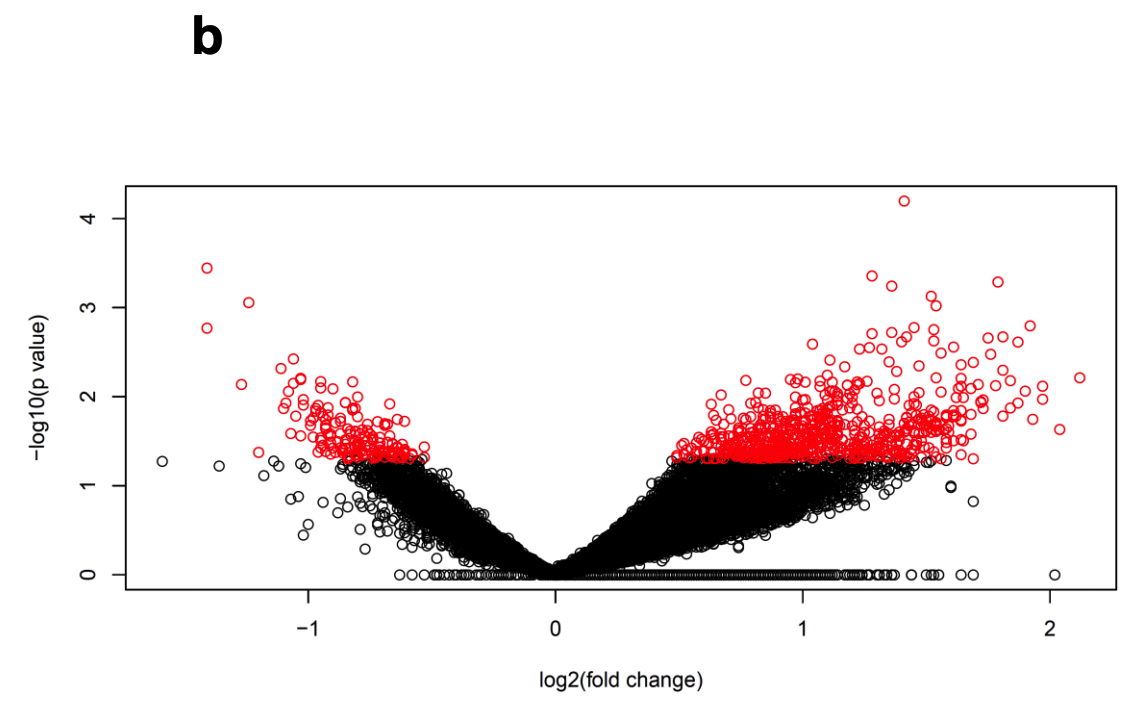
但是这并不影响研究者进行后续差异分析; (b) Volkano plot of human islet ATAC-seq data of donors with type 2 diabetes versus non-diabetic donors analysed with R Diffbind and edgeR packages- Interestingly, we found 1,078 differential ATAC-seq peaks in T2D versus control islets.
可以看到,符合要求的在糖尿病患者里面跟正常人相比有统计学显著的差异的ATAC-seq peaks仍然是有一千多个!
火山图有点丑:
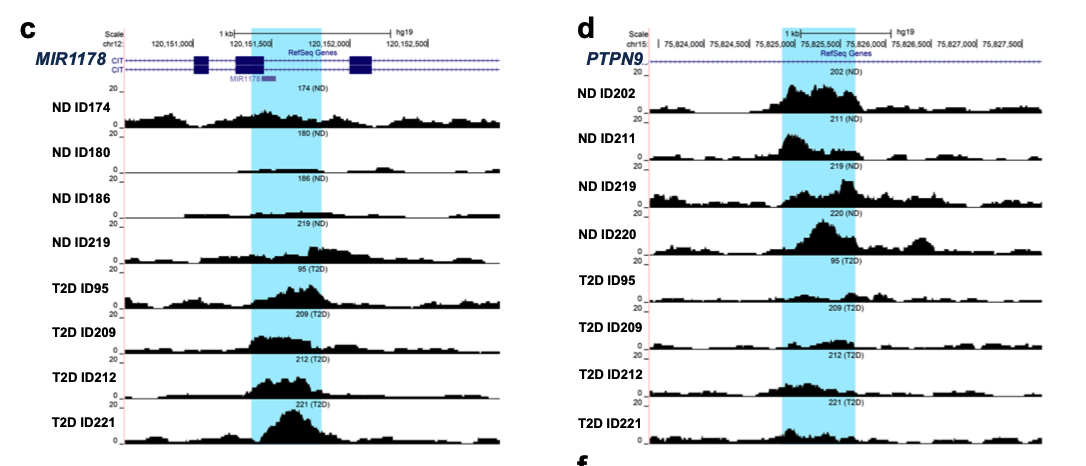
作者在糖尿病的病人组和正常对照组里面各自挑选了一个信号值改变的peak进行IGV的载入bw文件的可视化,如下所示:
可以看到,这个时候研究者很鸡贼哦,他们并没有去可视化这些peaks在全部的15个样品的ATAC-seq数据的差异,而是选择了4个糖尿病患者和4个正常对照,确实就很容易看到信号值的上下调!
如果是转录组数据差异分析,基本上看我六年前的表达芯片的公共数据库挖掘系列推文即可;- 解读GEO数据存放规律及下载,一文就够
- 解读SRA数据库规律一文就够
- 从GEO数据库下载得到表达矩阵 一文就够
- GSEA分析一文就够(单机版+R语言版)
- 根据分组信息做差异分析- 这个一文不够的
- 差异分析得到的结果注释一文就够
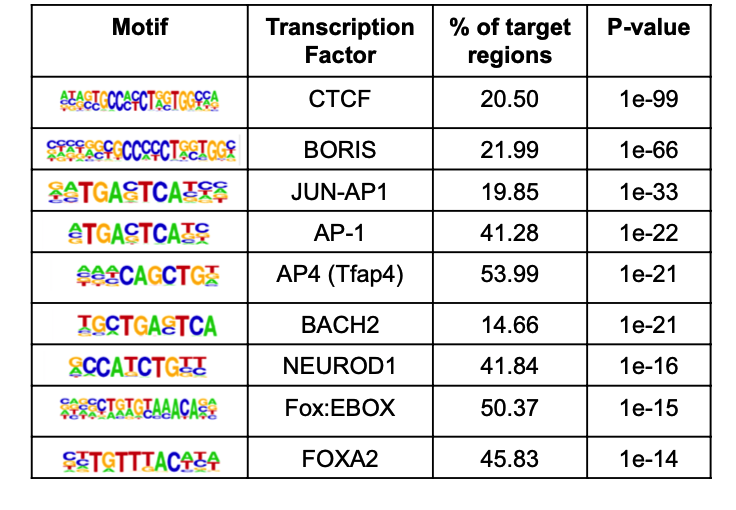
但这个文章做的是 ATAC-seq数据,差异分析首先就不一样,拿到了1,078 ATAC-seq peaks 虽然说可以对应到具体的基因,然后进行生物学功能数据库注释,但是比较有特色的分析应该是peaks的注释。
比如研究者,就针对在《糖尿病的病人组》里面上调的1,078 ATAC-seq peaks 进行HOMER的注释:
上游分析这里我略过了,感兴趣可以去看教程ATAC-seq项目的标准分析仅收费1600, 有一个2020综述《From reads to insight: a hitchhiker’s guide to ATAC-seq data analysis》值得看:- MACS2 进行 Peak calleing
- csaw 进行差异 Peak 分析
- MEME suite 进行 motif 检测和富集
- ChIPseeker 进行注释和可视化
- HMMRATAC 进行核小体检测
- HINT-ATAC 进行足迹分析
我们以 seurat 官方教程为例:
提起生物信息学灌水大家首先想到的肯定是我们中国特色的临床医师畸形科研现状,不过最近我看到了一个有意思的文章:《Integrated bioinformatic analysis identifies UBE2Q1 as a potential prognostic marker for high grade serous ovarian cancer》,如下所示: Continue reading