时隔好几个月,因为各种各样的原因数据终于拿到了自己的手上,真是不容易啊!
拿到数据后,第一件要做的事情就是检查数据传输的完整性,然后备份!我拿到的数据如下:
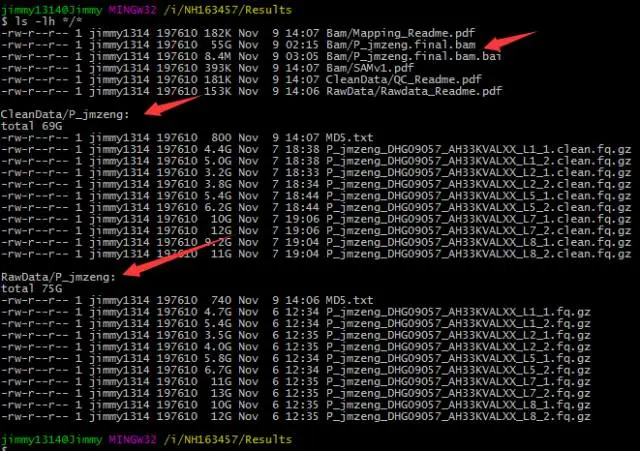
可以看到,公司给了我测序仪的下机数据(raw data)和他们质控后的clean data,这个过程减少了6G的数据量,对应着约90亿bp的碱基,相当于减少了3个人的全基因组数据。具体推算公式见前面的系列直播贴!
首先我把数据拷贝到了我上上周买的2T移动硬盘里面,再拷贝到我工作电脑一份,服务器一份,私人电脑一份,另外一个移动硬盘一份。然后删除了公司寄给我的硬盘里面的数据,再把硬盘寄回给公司,然后监督他们删除我所有的数据。(做这么多就是为了保护隐私,当然这个大前提是我已经确定数据没有问题了。)
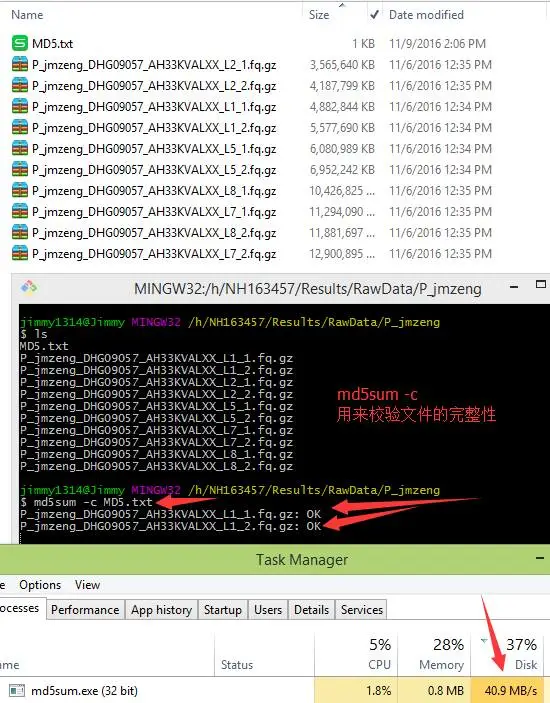
检查数据传输的完整性就是md5校验,看看数据在拷贝过程中有没有意外的损坏(这个在之前下载数据的时候我也说过)!一般传输数据之前,会用md5命令来生成各个文件的md5值,就是下面的MD5.txt文件里面的内容,然后传输数据之后,需要自行用md5sum -c MD5.txt 来校验文件里面记录的文件的完整性,如果显示都是OK,说明文件拷贝传输过程是没有问题的!但这个过程会耗费大量的磁盘读写,磁盘读写能力是有限的,所以开多个进程并不能加快这一过程。
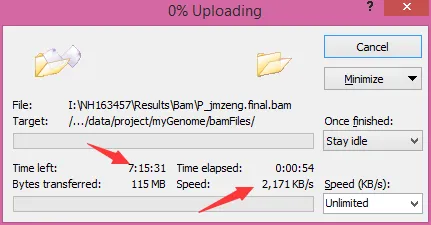
然后我把公司处理好的bam文件上传到服务器做下游分析,我用的winscp软件把文件传到服务器上的!
从明天起,我们就开始正式对基因组进行分析啦!欢迎围观!
请扫描以下二维码关注我们,获取直播系列的所有帖子!
