那么我们一起来看看科研及临床一般都是进行怎样的分析吧! Continue reading
十
26
那么我们一起来看看科研及临床一般都是进行怎样的分析吧! Continue reading
This online tool currently uses CrossMap, which supports a limited number of formats (see our online documentation for details of the individual data formats listed below). CrossMap also discards metadata in files, so track definitions, etc, will be lost on conversion.
Important note: CrossMap converts WIG files to BedGraph internally for efficiency, and also outputs them in BedGraph format.
X-DAS-Version: DAS/0.95 X-DAS-Status: 200 Content-Type:text Access-Control-Allow-Origin: * Access-Control-Expose-Headers: X-DAS-Version X-DAS-Status X-DAS-Capabilities UCSC DAS Server. See http://www.biodas.org for more info on DAS. Try http://genome.ucsc.edu/cgi-bin/das/dsn for a list of databases. See our DAS FAQ (http://genome.ucsc.edu/FAQ/FAQdownloads#download23) for more information. Alternatively, we also provide query capability through our MySQL server; please see our FAQ for details (http://genome.ucsc.edu/FAQ/FAQdownloads#download29). Note that DAS is an inefficient protocol which does not support all types of annotation in our database. We recommend you access the UCSC database by downloading the tab-separated files in the downloads section (http://hgdownload.cse.ucsc.edu/downloads.html) or by using the Table Browser (http://genome.ucsc.edu/cgi-bin/hgTables) instead of DAS in most circumstances.
今天先 对7个单端数据做处理,是454数据,平均长度300bp左右,明天再处理3KB和20KB的配对reads。
首先跑fastqc
打开一个个看结果
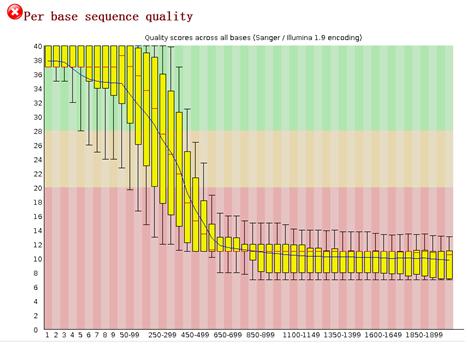
可以看到前面一些碱基的质量还是不错的, 因为这是454平台测序数据,序列片段长度差异很大,一般前四百个bp的碱基质量还是不错的,太长了的测序片段也不可靠
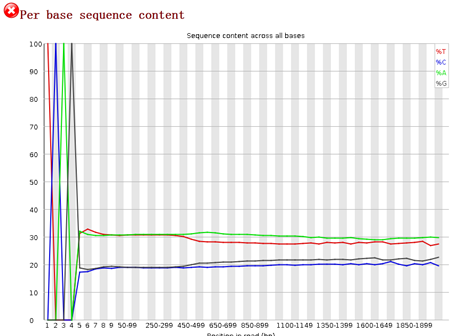
重点在下面这个图片,可以看到,前面的4个碱基是adaptor,肯定是要去除的,不是我们的测序数据。是TCAG,需要去除掉。
所以我们用了 solexaQA 这个套装软件对原始测序数据进行过滤
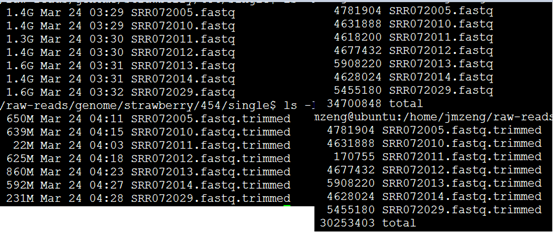
可以看到过滤的非常明显!!!甚至有个样本基本全军覆没了!然后我查看了我的批处理脚本,发现可能是perl DynamicTrim.pl -454 $id这个参数有问题
for id in *fastq
do
echo $id
perl DynamicTrim.pl -454 $id
done
for id in *trimmed
do
echo $id
perl LengthSort.pl $id
done
可以看到末尾的质量差的碱基都被去掉了,但是头部的TCAG还是没有去掉。
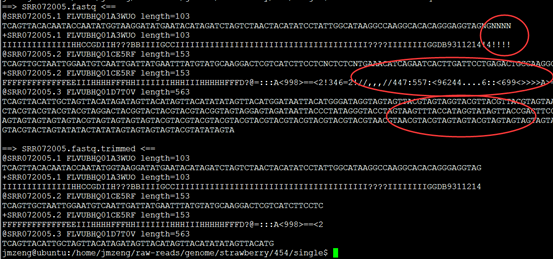
处理完毕后的数据如下:
找橡胶测序数据无果
所以我只好找了他们所参考的草莓(strawberry, Fragaria vesca (2n = 2x = 14),a small genome (240 Mb),)的文章,是发表是nature genetics上面的
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3326587/
可以看到它的SRA索取号。

草莓组装结果:Over 3,200 scaffolds were assembled with an N50 of 1.3 Mb .
Over 95% (209.8 Mb) of the total sequence is represented in 272 scaffolds.
草莓基因息:Gene prediction modeling identified 34,809 genes, with most being supported by transcriptome mapping.
草莓染色体信息:Paradoxically, the small basic (x = 7) genome size of the strawberry genus, ~240 Mb,
offers substantial advantages for genomic research.
草莓来源:diploid strawberry F. vesca ssp. vesca accession Hawaii 4
(National Clonal Germplasm Repository accession # PI551572).
然后我去NCBI上面下载这三个数据
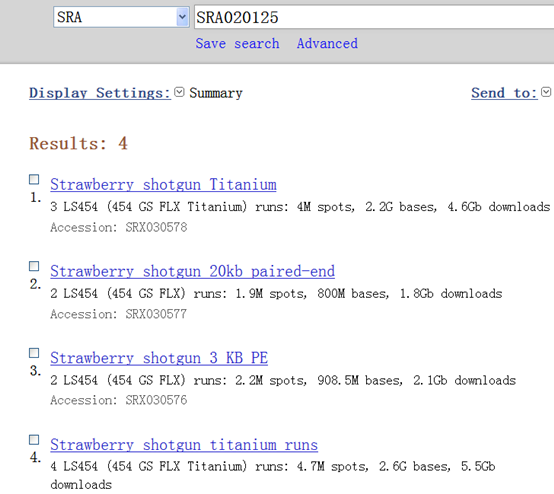
SRA020125 共有四个数据:
| http://www.ncbi.nlm.nih.gov/sra/SRX030575[accn] | Total: 4 runs, 4.7M spots, 2.6G bases, 5.5Gb |
| http://www.ncbi.nlm.nih.gov/sra/SRX030576[accn] (3 KB PE) | Total: 2 runs, 2.2M spots, 908.5M bases, 2.1Gb |
| http://www.ncbi.nlm.nih.gov/sra/SRX030577[accn] (20KB片段) | Total: 2 runs, 1.9M spots, 800M bases, 1.8Gb |
| http://www.ncbi.nlm.nih.gov/sra/SRX030578[accn] | Total: 3 runs, 4M spots, 2.2G bases, 4.6Gb |
挂在后台自动下载
![]()
好了,有了这些数据我们就要进行基因组的一系列分析啦!!!
不过我们可以先看看他们这个研究小组的成果
首先他们建造了一个关于草莓的基因组信息网站
https://strawberry.plantandfood.co.nz/
跟我之前在水科院做鲫鱼鲤鱼的差不多
直接在里面就可以下载他们做好的所有数据,也可以可视化。
它的染色体如下,非常简单,就七条染色体
http://www.rosaceae.org/species/fragaria/fragaria_vesca/genome_v1.1
我找到了它组装好的草莓基因组地址,用批处理全部下载了
