一个基因有一个甚至多个转录本!根据转录及翻译现象可以把一个基因人为的定义成多种结构!
首先自己查资料搞明白转录本,外显子和内含子,5端UTR区域和CDS区域,还有3端UTR区域的概念。
真核生物的基因含有外显子和内含子,是区别原核生物的特征之一,能转录的就是外显子,不能转录的就是内含子!
内含子(英语:Intron)是一个基因中非编码DNA片段,它分开相邻的外显子。更精确的定义是:内含子是阻断基因线性表达的序列。DNA上的内含子会被转录到前体RNA中,但RNA上的内含子会在RNA离开细胞核进行转译前被剪除。在成熟mRNA被保留下来的基因部分被称为外显子。
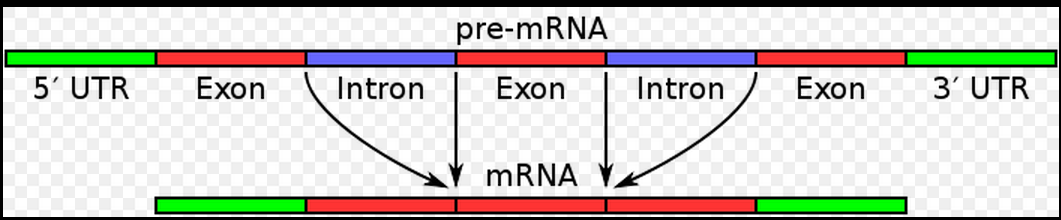
通常我们看到基因结构示意图会像下面这样!
但是对于这个图,我之前有非常多的疑问,直到我自己写程序去仔细统计,画图探究后才真正搞明白!
从图中可以看到5端UTR区域后面接着的是第一外显子,但是事实上不是!
外显子和内含子是转录本上面的概念,是基于转录这个行为的定义。
而5端UTR区域和CDS区域,还有3端UTR区域,是基于翻译这种行为的定义!
把它们画成这样,是严重的干扰信息。
实际上,我们需要更正很多概念,我检验了一下,得到下面几个正确的:
1,如果基因有多个转录本,基因的起始坐标,就是该基因所有转录本的第一个外显子的起始坐标的最小值,同理基因的终止坐标,就是该基因的所有转录本的最后一个外显子的终止坐标的最大值。
2,通过这个概念,可以把基因分成闭合基因和非闭合基因。 闭合基因:有一个最长转录本使得基因起始终止坐标等于该最长转录本的起始终止坐标。(这个是我乱说的,并没有这个定义)
3,如果基因只有一个转录本,那么基因的起始终止坐标,就是转录本的起始终止坐标!
4,一个基因的一个转录本的5'utr区域可以包括多个外显子区域,前者是翻译行为,后者是转录行为
5,起始密码子和终止密码子是CDS的起止处,是基于翻译的概念
6,一个基因的多个转录本的外显子坐标不一定会排列整齐,每个转录本的剪切位点并不一定要比其它转录本一致!
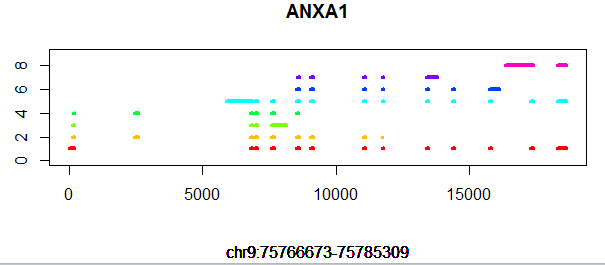
比如对这个ANXA1基因来说,非常多的转录本,但是基因的起始终止坐标,是所有转录本起始终止坐标的极大值和极小值!同时,它是一个闭合基因,因为它存在一个转录本的起始终止坐标等于该基因的起始终止坐标。可以看到它的外显子并不是非常整齐的,虽然多个转录本会共享某些外显子,但是也存在某些外显子比同区域其它外显子长的现象!
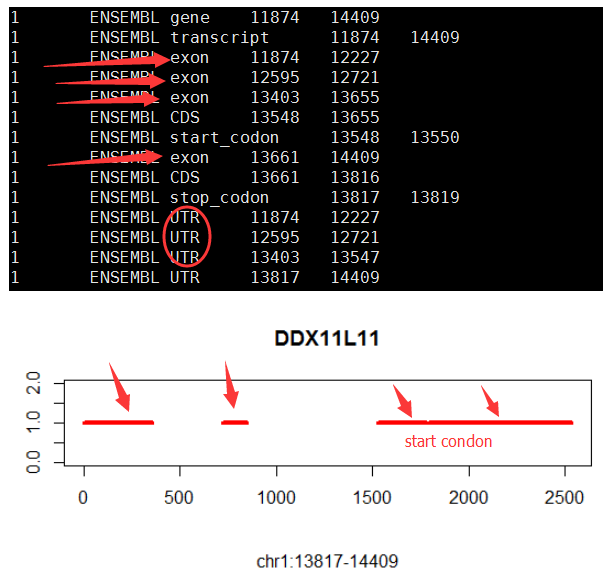
再比如下面这个例子:对DDX11L11这个基因来说,前两个外显子都不会翻译,直到第三个外显子才开始翻译,构成CDS。
有些转录本是没有utr的,所以该转录本的起始坐标,就是CDS的起始坐标
首先把gtf文件格式化导入mysql数据库
我用的是broadinstitute提供的GENCODE的gtf文件,是hg19版本的
##description: evidence-based annotation of the human genome (GRCh37), version 7 (Ensembl 62)
##provider: GENCODE
##format: gtf
##date: 2011-03-23
很简单,下载数据,parse好之后,用R导入mysql即可!
下面是mysql代码:
--mysql code
use test;
drop table if exists hg19_gtf;
create table hg19_gtf (
gene_name VARCHAR(30),
transcript_name VARCHAR(30) ,
record VARCHAR(15) NOT NULL ,
chr VARCHAR(2) NOT NULL ,
start INT NOT NULL ,
end INT NOT NULL ,
source VARCHAR(10) NOT NULL ,
strand VARCHAR(1) NOT NULL ,
gene_id VARCHAR(30) NOT NULL ,
transcript_id VARCHAR(30) NOT NULL ,
gene_status VARCHAR(30) ,
gene_type VARCHAR(30) ,
transcript_type VARCHAR(30) ,
transcript_status VARCHAR(30)
);
--我的网页好像不支持mysql代码高亮,大家凑合着看吧,反正就是一个简单的建表语句!
select * from hg19_gtf limit 100;
select * from hg19_gtf where gene_name='DMD';
select count(*) from hg19_gtf where gene_name='DMD' and record='start_codon'; --18 start condon
select count(distinct(transcript_name)) from hg19_gtf where gene_name='DMD' ; --34 transcript
select count(distinct(transcript_name)) c ,gene_name from hg19_gtf where record='transcript' group by gene_name order by c desc;
我是随意设计的一个表,主要是为了画图方便!
接下来是R code来具体的对基因进行画图!
[perl]
suppressMessages(library(ggplot2))
suppressMessages(library(RMySQL))
con <- dbConnect(MySQL(), host="127.0.0.1", port=3306, user="root", password="11111111")
dbSendQuery(con, "USE test")
gene='SOX10'
#gene='DDX11L11'
if (T){
query=paste("select * from hg19_gtf where gene_type='protein_coding' and gene_name=",shQuote(gene),sep="")
structure=dbGetQuery(con,query)
tmp_min=min(c(structure$start,structure$end))
structure$new_start=structure$start-tmp_min
structure$new_end=structure$end-tmp_min
tmp_max=max(c(structure$new_start,structure$new_end))
num_transcripts=nrow(structure[structure$record=='transcript',])
tmp_color=rainbow(num_transcripts)
x=1:tmp_max;y=rep(num_transcripts,length(x))
#x=10000:17000;y=rep(num_transcripts,length(x))
plot(x,y,type = 'n',xlab='',ylab = '',ylim = c(0,num_transcripts+1))
title(main = gene,sub = paste("chr",tmp$chr,":",tmp$start,"-",tmp$end,sep=""))
j=0;
tmp_legend=c()
for (i in 1:nrow(structure)){
tmp=structure[i,]
if(tmp$record == 'transcript'){
j=j+1
tmp_legend=c(tmp_legend,paste("chr",tmp$chr,":",tmp$start,"-",tmp$end,sep=""))
}
if(tmp$record == 'exon') lines(c(tmp$new_start,tmp$new_end),c(j,j),col=tmp_color[j],lwd=4)
}
# legend('topleft',legend=tmp_legend,lty=1,lwd = 4,col = tmp_color);
}
[/perl]
通过这个代码,,就能简单的得到我上面显示的基因结构图啦!