是福建医科大学的学者开发的,
文章里面详细讲解了他们的这个差异分析的统计学原理
大意就是找到同一组织的normal样本的表达量数据,几百个,这样就可以分析2万基因之间的互相配对,检测表达量是否在几百个样本里面稳定的不一样!
我现在还不是很确定这个方法,只是试一试,欢迎与我交流对该方法的讨论!
文章是:
Wang H, Sun Q, Zhao W, et al. Individual-level analysis of differential expression of genes and pathways for personalized medicine[J]. Bioinformatics, 2014: btu522.
他们把它写成了一个R包,可以下载使用,但是必须用R2.15.2版本,我用了一下,不好用!
We can download the R code for in http://bioinformatics.oxfordjournals.org/content/31/1/62/suppl/DC1
他们这个程序真心不好用,但是很容易看懂算法,可以自己用R语言写一个来实现同样的过程!
比如A基因在几百个样本里面表达量都是3左右,而B基因都是5左右,而且满足99%的A表达量高于B,那么这就是一个稳定的基因对!
一般2万基因之间可以配成2亿个基因对,其中稳定的大概有10%~40%
然后我们对每个疾病样本都可以进行检验,看看这样稳定的基因对是否被改变!
比如,我们拿到一个疾病样本的2万个基因的表达量,我们挑取一个,如果它有100个稳定的up的基因对,100个稳定的down的基因对
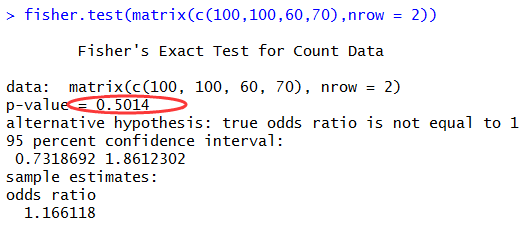
那么,我看看这些基因对是否被改变了,如果这样还有70基因对在该疾病样本里面仍然是up,60个是down,那么我用Fisher精确检验的结果是
这个基因在该疾病样本,相对于normal pool并没有差异表达!当然检验得到的P值最后可以做FDR校验。
依次这样,把所有的gene都分析完,就知道这个样本有哪些差异的gene了。
介绍完原理,我们拿一个具体的例子来看看吧:
首先我们下载一个2008年的一个人的肝脏表达数据(Gene Expression in Human Liver),都是正常组织,共427个样本。
不过这个芯片比较小众,是默克医药公司定制化的, 需要下载探针对应gene的文件!
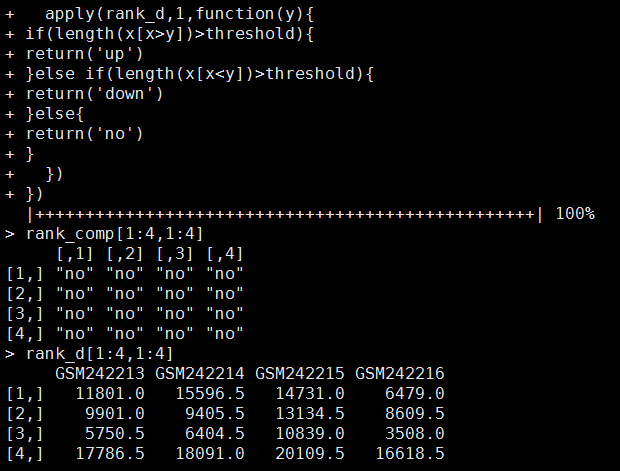
我们读取GSE9588这个数据,得到表达矩阵,然后计算rank矩阵,然后计算得到comp矩阵
> table(rank_comp)
rank_comp
down no up
58479 465752098 58479
>
不知道为什么这个数据,stable的那么少,不知道是不是出了什么问题!
其实我的程序都是对的了,只是因为这个数据集已经不是纯粹的表达量数据了,而是这427个样本的数据都减去了某个样本的表达量。
这样每个个体的基因之间的表达量排序就会被干扰,导致得到的稳定基因对非常少!!!
但是,我后来下载了GTEx的表达数据,拿那里面的normal组织样本表达量来做,可以得到非常多的稳定基因对。
实际代码大概是:
得到正常组织的表达矩阵:
然后计算表达矩阵的rank,得到各个样本自己的基因排序情况,得到排序矩阵!
处理排序矩阵,每个基因对之间都算一下是否稳定,得到稳定性描述矩阵!
然后根据每个疾病个体的基因表达情况,来循环每个基因, 看看该基因是否差异!