前些天逛bioStar论坛的时候看到了一个问题,是关于miRNA分析,提问者从NCBI的SRA数据下载文献提供的原始数据,然后处理的时候有些不懂,我看到他列出的数据是iron torrent测序仪的,而且我以前还没玩过miRNA-seq的数据分析, 就抽空自学了一下。因为我有RNA-seq的基础,所以理解学习起来比较简单。特记录一下自己的学习过程,希望对后学者有帮助。 Continue reading
六
25
前些天逛bioStar论坛的时候看到了一个问题,是关于miRNA分析,提问者从NCBI的SRA数据下载文献提供的原始数据,然后处理的时候有些不懂,我看到他列出的数据是iron torrent测序仪的,而且我以前还没玩过miRNA-seq的数据分析, 就抽空自学了一下。因为我有RNA-seq的基础,所以理解学习起来比较简单。特记录一下自己的学习过程,希望对后学者有帮助。 Continue reading
文献:Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes
A small pre-defined set of gene expression signatures
| epithelial-to-mesenchymal transition (EMT) | 上皮细胞向间充质细胞转化 |
| microsatellite instability (MSI) | 微卫星不稳定性 |
| cytokine signaling | 细胞因子信号 |
| cell proliferation | 细胞增殖 |
| DNA methylation | DNA甲基化 |
| TP53 activity | TP53活性 |
| gastric tissue | 胃组织 |
经典的分类方法是:Gastric cancer may be subdivided into 3 distinct subtypes—proximal, diffuse, and distal gastric cancer—based on histopathologic and anatomic criteria. Each subtype is associated with unique epidemiology.
我们用主成分分析Principal component anaylsis (PCA)
PC1
PC2
PC3
这三个主成分与上面的七个特征是相关联的。
根据我们的主成分分析,可以把我们的300个GC样本分成如下四组,命名如下:
Gene expression signatures define four molecular subtypes of GC:
MSI (n = 68),
MSS/EMT (n = 46),
MSS/TP53+ (n = 79)
MSS/TP53− (n = 107)
然后用本文的分类方法,测试了另外另个published数据,还是分成四个组
(MSI, MSS/EMT, MSS/TP53+ and MSS/TP53-)
分别是TCGA数据库的;n = 46, n = 62, n = 50 and n = 47.
Singapore的研究; n = 12, n = 85, n = 39 and n = 63 respectively
我们这样的分组可以得到一些规律:
(i) The MSS/EMT subtype occurred at a significantly younger age (P = 3e-2) than did other subtypes. The majority (>80%) of the subjects in this subtype were diagnosed with diffuse-type (P < 1e-4) at stage III/IV(P = 1e-3).
(ii) The MSI subtype occurred predominantly in the antrum (75%), >60% of subjects had the intestinal subtype, and >50% of subjects were diagnosed at an early stage (I/II).
(iii) Epstein-Barr virus (EBV) infection occurred more frequently in the MSS/TP53+ group (n = 12/18, P = 2e-4) than in the other groups.
然后我们对我们的300个样本做了生存分析:
预后: MSI > MSS/TP53+ > MSS/TP53 > MSS/EMT
Next, we validated the survival trend of GC subtypes in three independent cohorts: Samsung Medical Center cohort 2 (SMC-2,n = 277, GSE26253)31,
Singapore cohort(n = 200, GSE15459)21 and
TCGA gastric cohort (n = 205).
We saw that the GC subtypes showed a significant association with overall survival
结论:我们这样的分类是最合理的,跟各个类别的预后非常相关。
然后我们看看突变模式:
the MSI~ hypermutation ~KRAS (23.3%), the PI3K-PTEN-mTOR pathway (42%), ALK (16.3%) and ARID1A (44.2%)18.
We observed enrichment of PIK3CA H1047R mutations in the MSI samples
we saw enrichment of E542K and E545K mutations in MSS tumors
The EMT subtype had a lower number of mutation events when compared to the other MSS groups(P = 1e−3).
The MSS/TP53− subtype showed the highest prevalence of TP53 mutations (60%), with a low frequency of other mutations
the MSS/TP53+ subtype showed a relatively higher prevalence (compared to MSS/TP53−) of mutations in APC, ARID1A, KRAS, PIK3CA and SMAD4.
再看看拷贝数变异情况:
再看看与另外两个研究团队的分类情况的比较
The TCGA study reported expression clusters (subtypes named C1–C4) and genomic subtypes (subtypes named EBV+, MSI, Genome Stable (GS) and Chromosomal Instability (CIN)).
A follow-up study of the Singapore cohort21 described three expression subtypes (Proliferative, Metabolic and Reactive)
However, a consensus on clinically relevant subtypes that encompasses molecular heterogeneity and that can be used in preclinical and clinical research has not been reported.
Here we report the molecular classification of GC linked not only to distinct patterns of genomic alterations, but also to recurrence pattern and prognosis across multiple GC cohorts.
microsatellite instability
英文简称 : MI
中文全称 : 微卫星不稳定性
所属分类 : 生物科学
词条简介 : 微卫星不稳定性(microsatellite instability,MI)检测是基于VNTR的发现,细胞内基因组含有大量的碱基重复序列,一般将6-7bp的串联重称为小卫星DNA(minisatellite DNA),又称为VNTR。而将1-4bp的串联重复称为微卫星DNA,又称简单重复序列(simple repeat sequence,SRS)。SRS是一种最常见的重复序列之一,具有丰富的多态性、高度杂合性、重组纺低等优点。最常见的为双核苷酸重复,即(AC)n和(TG)n。研究表胆,在n≥104时,2bp重复序列在人群中呈高度多态性。SRS广泛存在于原核和真核基因组中,约占真核基因组的5%,是近年来快速发展起来的新的DNA多态性标志之一。策卫星稳定性(MI)是指简重复序列的增加或丢失。MI首先在结肠癌中观察到,1993年在HNPCC中观察到多条染色体均有(AC)n重复序列的增加或毛失,以后相继在胃癌、胰腺癌、肺癌、膀胱癌、乳腺癌、前列腺癌及其他肿瘤等也好现存在微卫星不稳定现象,提示MI可能是肿瘤细胞的另一重要分子结果显示 ,MI与肿瘤与发展有关,MI仅在肿瘤细胞中发现,从未在正常组织中检测到。在原发与移肿瘤中,MI均交分布于整个肿瘤。晚期胃癌的MI频率显著高于早期胃癌。
研读橡胶的基因组文章
我本科的前两年在海南儋州读书,那时候旁边就是橡胶所,很多同学也在那边做毕业论文什么的,我一直以为那里是全世界的橡胶中心,所有的先进技术都在那里产生,结果,前些天跟一个橡胶所的老师聊天才发现,居然橡胶(Hevea brasiliensis)的基因组已经发表了,可是,跟橡胶所没有半毛钱关系,更搞笑的事情是,堂堂一个基因组文章居然发表在BMC这样的杂志,真不知道是基因组的年代已经过去了还是他们做的实在是太差了,反正我看不过去了,所以研读他们的文章,并且下载数据测试一下。
文章地址如下:http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3575267/

可以看到它过于数据的描述都在补充材料1里面,所以我下载了补充材料。
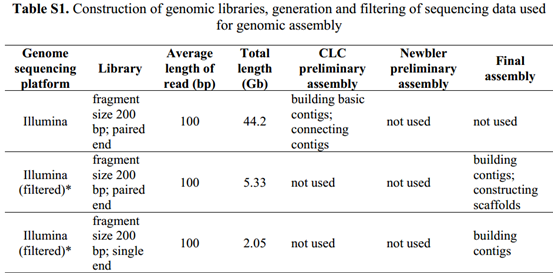
可以看到所有的测序数据的描述,45个G的i llumina的200bp的双端测序,27个G的illumina的200bp的双端测序,约10G左右的长片段(8kb,20kb)罗氏454数据,最后还有一点点solid数据,它这样的测序策略好像是模仿的2011年发布的草莓基因组数据。
但是补充材料里面没有列出下载地址,我有点困惑!
按照道理我研读文献的步骤应该没有错,有可能是因为这个文章发表的杂志水平太低,所以不要求他们把测序原始数据上传到NCBI的SRA里面。或者是他们本身觉得文章发的不够档次,不想公布数据,所以先留着自己做精细分析,等发了大文章再公布原始数据。
然后我在NCBI的SRA里面查找了关于橡胶的原始数据,果真没有
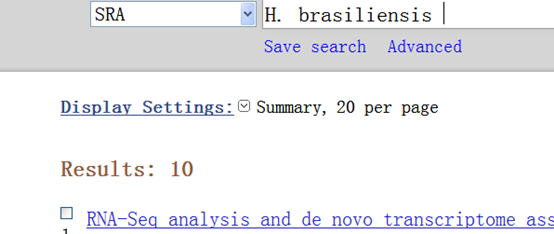
仅有的10个数据,都是别的小组做的RNA-seq的内容。
De novo transcriptome analysis of abiotic stress responsive transcripts of Hevea brasiliensis.
所以我只好找了他们所参考的草莓(strawberry, Fragaria vesca (2n = 2x = 14),a small genome (240 Mb),)的文章,是发表是nature genetics上面的