上一次直播中,我们对拿到手的测序数据进行了质控,测序数据的质量已经得到了保证。那么接下来就可以把它拿来与参考基因组比对了,这里我们先用参考基因组hg19,大家可以参照【直播】我的基因组(五):测试数据及参考基因组的准备来下载参考基因组hg19,我这里选择的是UCSC提供的hg19。然后安装bwa软件进行比对,可以参考【直播】我的基因组(四):计算资源的准备来安装,以及对hg19建立索引。 Continue reading
十二
09
上一次直播中,我们对拿到手的测序数据进行了质控,测序数据的质量已经得到了保证。那么接下来就可以把它拿来与参考基因组比对了,这里我们先用参考基因组hg19,大家可以参照【直播】我的基因组(五):测试数据及参考基因组的准备来下载参考基因组hg19,我这里选择的是UCSC提供的hg19。然后安装bwa软件进行比对,可以参考【直播】我的基因组(四):计算资源的准备来安装,以及对hg19建立索引。 Continue reading
很多时候,我们都要选取unique mapped的reads,尤其是在RNA-seq和CHIP-seq的时候,但是如何保留,各种教程都不一致,我稍微总结了一下,是因为使用的比对工具不一样导致的!但是主要都反应在sam文件的一系列tag里面~
首先对bwa来说,如果它遇到一个reads可以比对到参考基因在的多个序列,只会随机的选取一个位置来输出到sam文件,但是会加上一个tag是XS:I:<N>来告诉我们第二好的比对情况的比对得分是多少,bowtie也是一样。但是它们都有参数来决定是否只对每个reads输出一条信息,还是输出全部的信息,在bwa是-a的参数,在bowtie里面是-m参数。
但是bowtie2里面取消了这个参数,它们都必须用XS:I:<N>这个tag来挑选unique mapped的reads
但是如果是用hisat来比对的话,决定是否是唯一比对的却是NH这个tag信息。默认情况下一条reads可以输出多条比对结果。
我想起了再补充吧,其实应该找几个例子用IGV看看,就明白了,可是我暂时没有时间了,只是觉得这个很重要,就提一下。
0=blue, 1=green, 2=orange, 3=red) or as characters A/C/G/T (A=blue, C=green, G=orange, T=red).我们通常称为csfastq格式。也不知道是什么原因,对国产软件总是提不起兴趣,所以尽管SOAP系列都已经发展到了十几个软件了,我依然没有去试用一下。
# download a test reference genome (TAIR9 Chromosome 1)
wgethttp://biocluster.ucr.edu/~tbackman/query.fastq# download some test Illumina reads from Arabidopsis
运行命令:
2bwt-builder genome.fasta
# create binary of reference genome
soap -a query.fastq -D genome.fasta.index -o output.soap
# align query to genome and store output
结果解读:
tar jxvf software.tar.bz2cd software./configure --prefix=$pathmakemake test
# download a test reference genome (TAIR9 Chromosome 1)
wgethttp://biocluster.ucr.edu/~tbackman/query.fastq# download some test Illumina reads from Arabidopsis
运行命令:
maq # inspect command line options
maq fasta2bfa genome.fasta genome.bfa
# create binary of reference genome
maq fastq2bfq query.fastq readBinary.bfq
# create a binary of dataset
maq match out.map genome.bfa readBinary.bfq
# align query to genome and store output
结果解读:
out.map肯定不是sam格式的。哈哈,这个软件我无法安装,换了好几系统也没成功,如果是太老了,很多库文件却是。我也懒得去解决了。这种报错,对我这样的非计算机专业来说,简直是天书!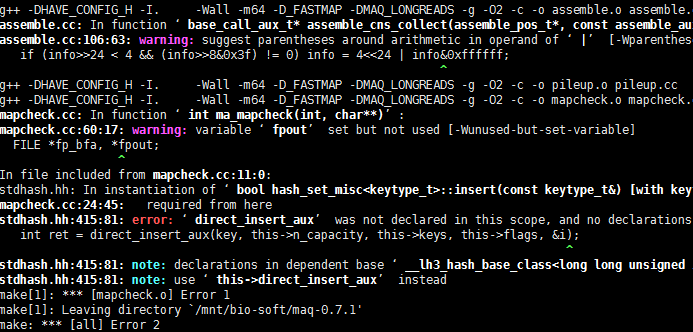
无意中看到了这个网站,比wiki的还有全面和专业。搜集了现有还算比较出名的比对软件,并且列出来了,还做了简单评价,里面对比对工具的收集,主要是基于2012年的一个综述《Tools for mapping high-throughput sequencing data》,相信应该是有不少人都看过这篇综述的,其实生物信息初学者应该自己去文献数据库找点感兴趣的关键词的综述多看看,广泛涉猎总没有坏处的。
<img src="http://www.ebi.ac.uk/~nf/hts_mappers/mappers_timeline.jpeg" alt="Mappers Timeline" width="800">
The following Table enables a comparison of mappers based on different characteristics. The table can be sorted by column (just click on the column name). The data was collected from different sources and in some cases was provided by the developers. For execution times and memory requirements we refer to the above mentioned review (supplementary data is available here).
软件的解说ppt :http://www.mi.fu-berlin.de/wiki/pub/ABI/CompMethodsWS11/MHuska_GSNAP.pdf
一.下载并且安装该软件
这是最新版本了
| Release 0.4.4 | 2012-10-30 | source code including documentation |
Wget http://1001genomes.org/data/software/genomemapper/genomemapper_0.4.4/genomemapper-0.4.4.tar.gz
这个软件安装很简单,解压进入目录,make一下即可
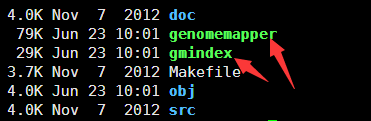
看到make完了之后就会多了两个软件,其中一个是用来构建参考基因组索引,一个用来比对的!
二.准备数据
既然是比对软件,那么肯定是一个参考基因组,一个测序的fastq原始文件咯
当然这个软件比较奇葩,它还支持Multi-FASTA, FASTQ2 or SHORE flat file format,
三、比对命令
这里要分两步走,首先是构建参考基因组的索引,然后才是比对
/home/jmzeng/bio-soft/genomemapper-0.4.4/gmindex \
-i BRCA1.fa -x BRCA1.idx -t BRCA1.meta
首先构建索引,种子长度就用默认的12即可,然后构建完索引如下。
然后进行比对即可
/home/jmzeng/bio-soft/genomemapper-0.4.4/genomemapper \
-i BRCA1.fa -q SRR258835.fastq -M 4 -G 2 -E 4 -o mapped_reads.fl -u unmapped_reads.fl
成功比对的都输出到了mapped_reads.fl -这个文件,未比对上的在unmapped_reads.fl
我有12344条序列,成功比对的只有5276条,但是如果我用精确比对的算法,只有一千五百条是可以比对的,所以用这个允许4个mismatch和2个gap的比对算法,大大提高了比对率。
然后我修改了比对参数可以达到5605,5654,5696的提升。但是没有质的飞跃,估计本身我的这种helicos测序数据错误率就太可怕了。
四,输出结果解读
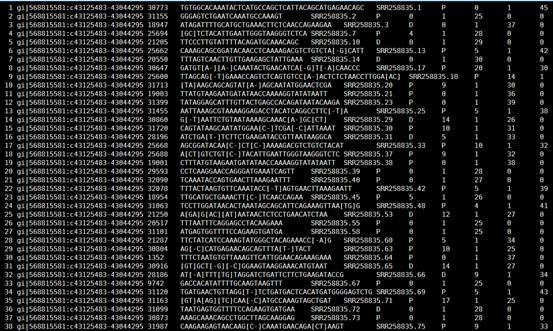
这个是很规则的tab键分割的文本字符,我就不解读了,大家看readme
一.下载并安装该软件
软件主页是http://samstat.sourceforge.net/ 里面对该软件进行非常详细的说明
包括installation和usage,我这里简单的翻译一下。
Wget http://liquidtelecom.dl.sourceforge.net/project/samstat/samstat-1.5.tar.gz
解压开看里面的readme有介绍如何安装这个软件
Unpack the tarball:
bash-3.1$ tar -zxvf samstat-XXX.tar.gz
bash-3.1$ cd samstat
bash-3.1$ ./configure
bash-3.1$ make
bash-3.1$ make check
bash-3.1$ make install
如果用root命令就可以直接用samstat啦
如果没有root权限,安装的时候稍微有点不同
./configure --prefix=/home/jmzeng/my-bin/
make
make install
很简单的
二,数据,就是我们的bam文件啦
三,运行命令
四,结果
简单看看samtools flagstat 740WT1.bam 的结果
19232378 + 0 in total (QC-passed reads + QC-failed reads)
0 + 0 duplicates
18846845 + 0 mapped (98.00%:-nan%)
0 + 0 paired in sequencing
0 + 0 read1
0 + 0 read2
0 + 0 properly paired (-nan%:-nan%)
0 + 0 with itself and mate mapped
0 + 0 singletons (-nan%:-nan%)
0 + 0 with mate mapped to a different chr
0 + 0 with mate mapped to a different chr (mapQ>=5)
然后再看看我们的samstat的结果!
740WT1.bam.samstat.html
一个网页,非常丰富的内容
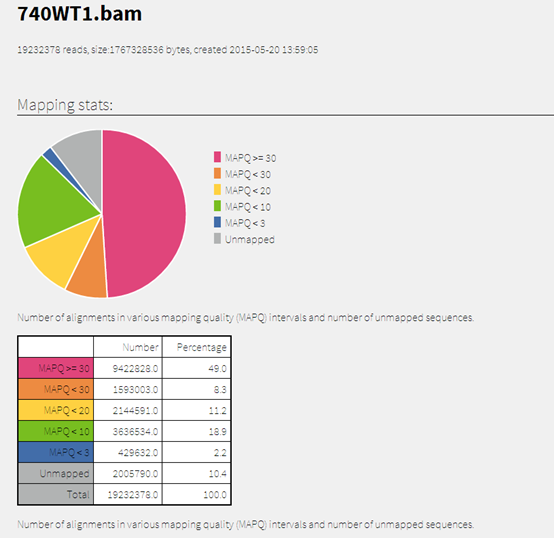
内容太多了,我懒得解释了
见软件说明书http://davetang.org/wiki/tiki-index.php?page=SAMStat
软件的主页是
进入主页,简单看看软件介绍,这个软件还是蛮牛的,一个人在家里自己写出来的,当然,对于普通人来说,这个软件跟clustalW没什么区别,反正都是多序列比对啦!
我们下载适合我们平台的版本即可!
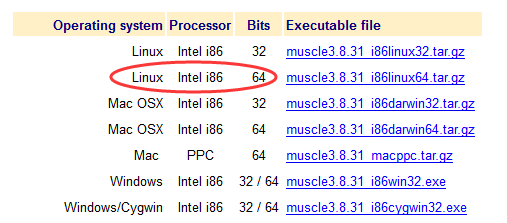
准备数据,我这里选择的是几个短小的蛋白
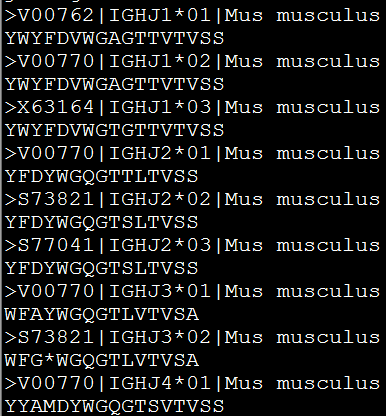
这里有两种比对方式,都是很简单的命令
一种是先比对,再生成树文件(树的格式是Newick format, )
muscle -in mouse_J.pro -out mouse_J.pro.a
muscle -maketree -in mouse_J.pro.a -out mouse_J.phy (这里有两种构建树的方式)
另外一种是比对成aln格式的数据,然后用其它软件(phyml或者phylip)来生出树文件
muscle -in mouse_J.pro -clwout seqs.aln
可以看到比对的效果还是蛮好的,是aln格式的比对文件,这个格式非常常用
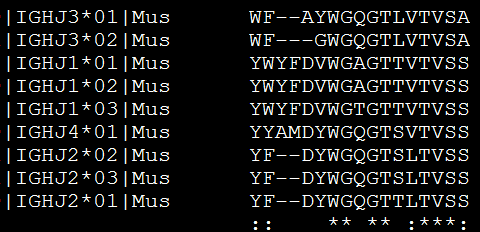
或者输出phy格式的比对文件,
muscle -in mouse_J.pro -physout seqs.phy
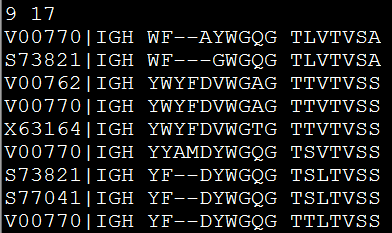
可以被phyml等软件识别,然后来构建进化树,见 http://www.bio-info-trainee.com/?p=626
下载地址我就不贴了,随便谷歌一下即可!
Genome Reference Consortium Human ---》 GRCh3
Feb. 2009 (hg19, GRCh37)这个是重点
Mar 2006 assembly = hg18 = NCBI36.
May 2004 assembly = hg17 = NCBI35.
July 2003 assembly = hg16 = NCBI34
以前的老版本就不用看啦,现在其实都已经有hg38出来啦,GRCh38 (NCBI) and hg38(UCSC)
参考:http://age.wang.blog.163.com/blog/static/119252448201092284725460/
http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/
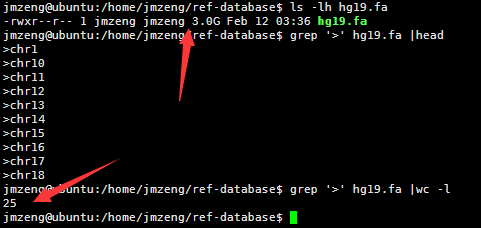
人的hg19基因组是3G的大小,因为一个英文字符是一个字节,所以也是30亿bp的碱基。
包括22条常染色体和X,Y性染色体及M线粒体染色体。
查看该文件可以看到,里面有很多的N,这是基因组里面未知的序列,用N占位,但是觉得部分都是A.T.C.G这样的字符,大小写都有,分别代表不同的意思。
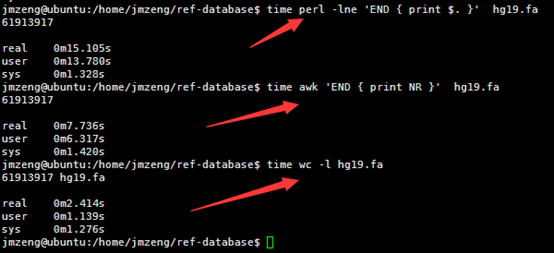
然后我用linux的命令统计了一下里面这个文件的行数,
perl -lne 'END { print $. }' hg19.fa
awk 'END { print NR }' hg19.fa
wc -l hg19.fa
然后我写了一个脚本统计每条染色体的长度,42秒钟完成任务!
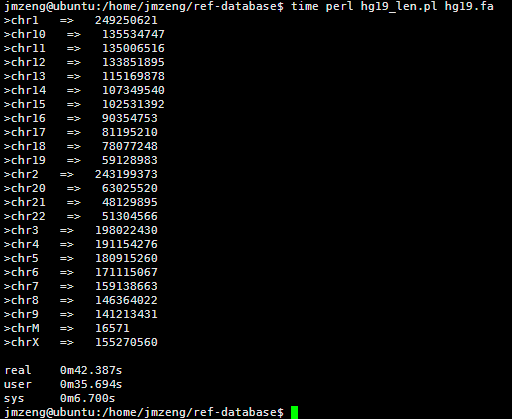
看来这个服务器的性能还是蛮强大的,读取文件非常快!
[perl]
while(<>){
chomp;
if (/>/){
if (exists $hash_chr{$key} ){
$len = length $hash_chr{$key};
print "$key => $len\n";
}
undef %hash_chr;
$key=$_;
}
else {
$hash_chr{$key}.=$_;
}
}
[/perl]
然后我用seed统计了一下hg19的词频(我不知道生物信息学里面的专业描述词语是什么)
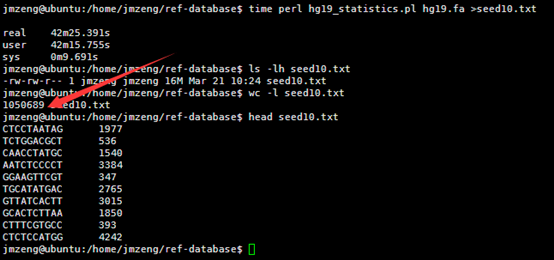
我的程序耗费了42分钟才跑完,感觉我写的程序应该是没有问题的,让我吃惊的是总共竟然只有105万条独特的10bp短序列。然后我算了一下4的10次方,(⊙o⊙)…悲剧,原来只有1048576,之所以出现这种情况,是因为里面有N这个字符串,不仅仅是A.T.C.G四个字符。我用grep -v N seed10.txt |wc -l命令再次统计了一下,发现居然就是1048576,也就是说,任意A.T.C.G四个字符组成的10bp字符串短序列在人的基因组里面都可以找到!!!
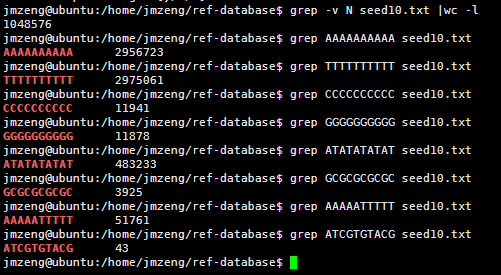
然后我测试了一下,还是真是这样的,真是一个蛮有意思的现象。虽然我无法解释为什么,但是根据这个结果我们可以得知连续的A或者T在人类基因组里面高频出现,而连续的G或者C却很少!
如果我们储存这个10bp字符串的同时,也储存着它们在基因组的位置,那么就可以根据这个seed来进行比对,这就是blast的原理之一!
首先进入bowtie的主页,千万要谷歌!!!
http://bowtie-bio.sourceforge.net/bowtie2/index.shtml
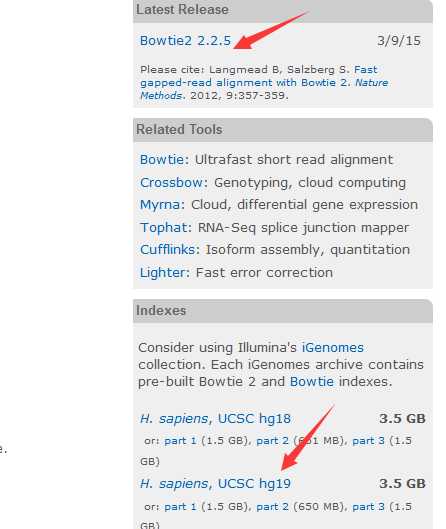
主页里面有下载链接,也有索引文件,当然索引文件只有人类等模式生物
下载之后是个压缩包,解压即可使用
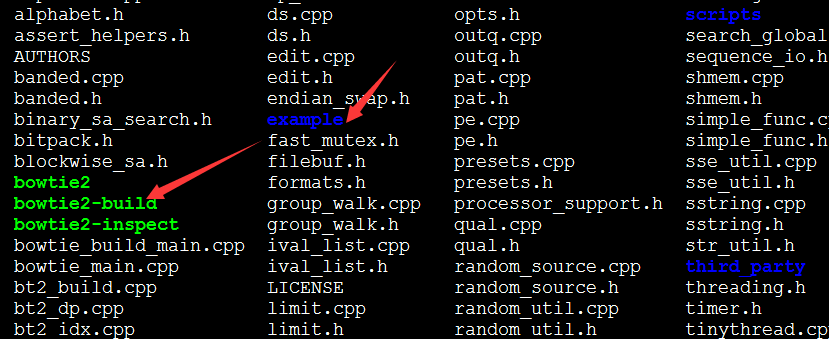
可以看到绿色的就是命令,可以添加到环境变量使用,也可以直接用全路径使用它!!!
然后example文件夹里面有所有的测试文件。
二、准备数据
我们就用软件自带的测试数据
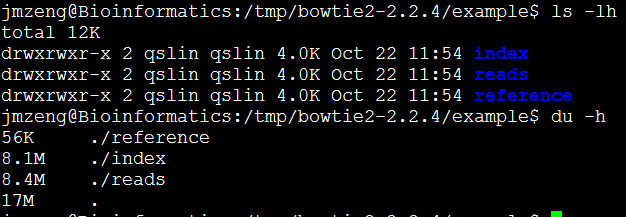
三、运行命令
分为两步,首先索引,然后比对!!!
然后你的目录就产生了六个索引文件,我给索引取名是tmp,你们可以随便取名字
重点参数的-x 和 –S ,单端是-U 双端是-1 -2
Bowtie –x tmp –U reads.fa –S hahahhha.sam
Bowtie –x tmp -1 reads1.fa -2 reads2.fa –S hahahha.sam
四:输出文件解读
就是输出了sam文件咯,这个就看我的sam文件格式讲解哈
转录组比对软件tophat的使用
为什么要用这个软件?:因为转录组reads比对到基因组reads用bwa和bowtie的效果都不够好,所以我们选择tophat
它做了什么?:tophat把测序的转录组的原始reads比对到了参考基因组上面,并且输出了bam(二进制的sam)文件比对结果给我们。(fastq--->bam)
一:下载安装该软件
其实一般的生信服务器自然会有高手给安装好了,你只需调用即可,这里我给大家演示一下如何安装。
wget http://ccb.jhu.edu/software/tophat/downloads/tophat-2.0.13.Linux_x86_64.tar.gz