查询需要根据前面建立的索引来做。
这是一个比较复杂的过程,我也是看了bowtie的作者的ppt才慢慢弄懂的,感觉自己也不可能三言两语就说清楚,一般都是辅助图片,动画,再经过多方交流才能慢慢理解。
所以大家呢,就自己去看ppt,看懂那个查询算法。(ppt及代码在我的群里面有共享,欢迎大家加群交流)
这里我简单讲讲我的程序
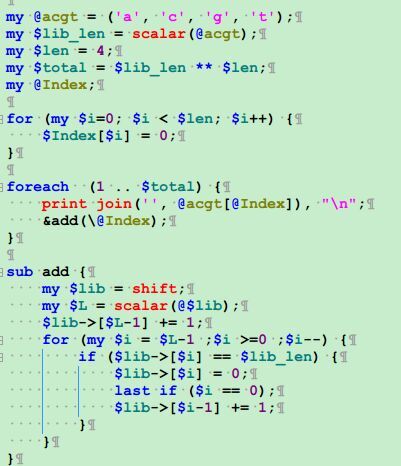
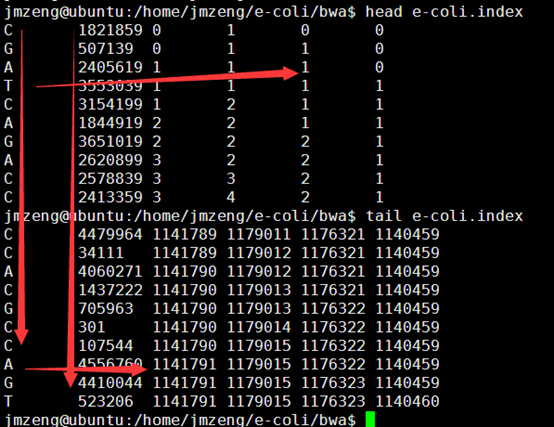
首先读取索引文件,统计好A,C,G,T的总数
然后把查询序列从最后一个字符往前面回溯。
我创建了一个子函数,专门来处理回溯的问题
每次接受四个参数(左右两端的碱基,上下的阈值),并返回两个参数(新的上下两个阈值)
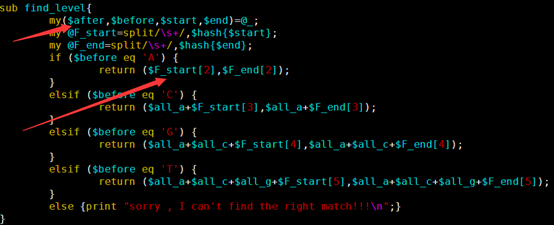
大家要看懂阈值是如何更新迭代,这样动态的一个个回溯字符串,一个个迭代阈值。
直到四种临界情况的出现。
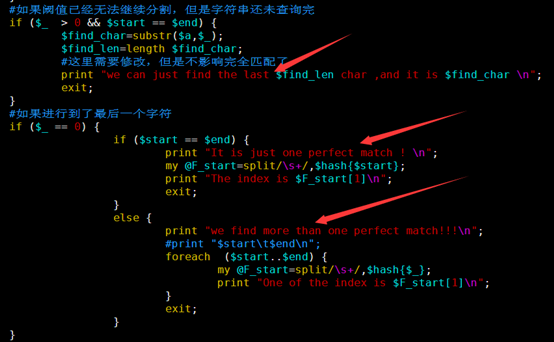
第一是上下阈值已经相等了,但是我们还没有回溯完全,那就说明字符串只能查找后几个字符,前面还有字符是无法匹配的
第二种情况是上下阈值已经相等了,正巧我们也回溯到了最后一个字符串,那么我们就找到了精确匹配。
第三种情况是已经进行到了最后一个字符串,但是上下阈值还有差值,那么就找到了多个精确匹配点。
最后一种情况是各种非法字符。
然后我简单的测序了一下在病毒的5K基因组里面的精确匹配情况,好像效果还挺好的
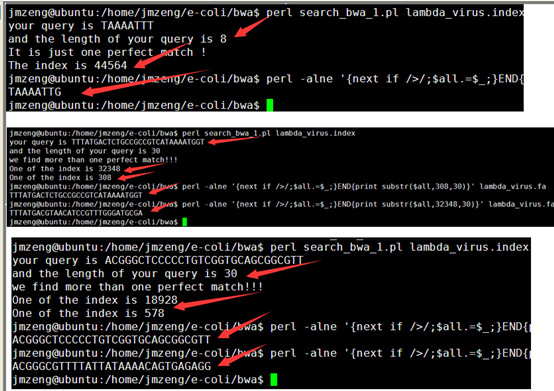
但是在酵母里面还有一个问题没有解决,就是取前二十个字符串排序的问题,不够精确,需要重新审视排序结果进行局部优化,可能是需要用堆排序发,具体我还得考虑一个星期,只能等下周上课再看看了,平时太忙了,基本没时间码代码。
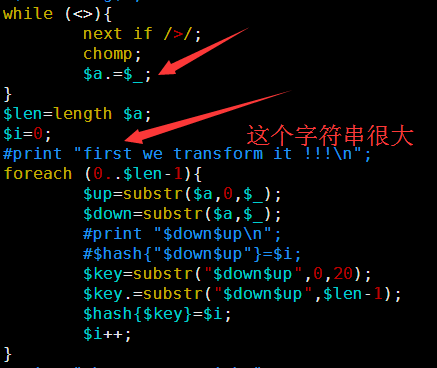
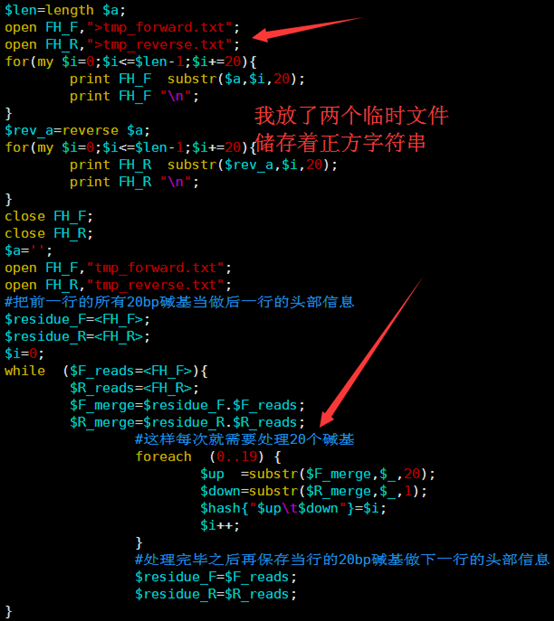
这里贴上我的代码给大家看看,
[perl]
$a='CGCTATGTACTGGATGCGCTGGCAAACGAGCCTGCCGTAAG';
while(<>){
chomp;
@F=split;
$hash_count_atcg{$F[0]}++;
$hash{$.}=$_;
}
$all_a=$hash_count_atcg{'A'};
$all_c=$hash_count_atcg{'C'};
$all_g=$hash_count_atcg{'G'};
$all_t=$hash_count_atcg{'T'};
#print "$all_a\t$all_c\t$all_g\t$all_t\n";
$len_a=length $a;
$end_a=$len_a-1;
print "your query is $a\n";
print "and the length of your query is $len_a \n";
foreach (reverse (0..$end_a)){
$after=substr($a,$_,1);
$before=substr($a,$_-1,1);
#对第一个字符进行找阈值的时候,我们需要人为的定义起始点!
if($_ == $end_a){
if ($after eq 'A') {
$start=1;
$end=$all_a;
}
elsif ($after eq 'C') {
$start=$all_a+1;
$end=$all_a+$all_c;
}
elsif ($after eq 'G') {
$start=$all_a+$all_c+1;
$end=$all_a+$all_c+$all_g;
}
elsif ($after eq 'T'){
$start=$all_a+$all_c+$all_g+1;
$end=$all_a+$all_c+$all_g+$all_t;
}
else {print "error !!! we just need A T C G !!!\n";exit;}
}
#如果阈值已经无法继续分割,但是字符串还未查询完
if ($_ > 0 && $start == $end) {
$find_char=substr($a,$_);
$find_len=length $find_char;
#这里需要修改,但是不影响完全匹配了
print "we can just find the last $find_len char ,and it is $find_char \n";
exit;
}
#如果进行到了最后一个字符
if ($_ == 0) {
if ($start == $end) {
print "It is just one perfect match ! \n";
my @F_start=split/\s+/,$hash{$start};
print "The index is $F_start[1]\n";
exit;
}
else {
print "we find more than one perfect match!!!\n";
#print "$start\t$end\n";
foreach ($start..$end) {
my @F_start=split/\s+/,$hash{$_};
print "One of the index is $F_start[1]\n";
}
exit;
}
}
($start,$end)=&find_level($after,$before,$start,$end);
}
sub find_level{
my($after,$before,$start,$end)=@_;
my @F_start=split/\s+/,$hash{$start};
my @F_end=split/\s+/,$hash{$end};
if ($before eq 'A') {
return ($F_start[2],$F_end[2]);
}
elsif ($before eq 'C') {
return ($all_a+$F_start[3],$all_a+$F_end[3]);
}
elsif ($before eq 'G') {
return ($all_a+$all_c+$F_start[4],$all_a+$all_c+$F_end[4]);
}
elsif ($before eq 'T') {
return ($all_a+$all_c+$all_g+$F_start[5],$all_a+$all_c+$all_g+$F_end[5]);
}
else {print "sorry , I can't find the right match!!!\n";}
}
#perl -alne '{next if />/;$all.=$_;}END{print substr($all,308,10)}' lambda_virus.fa
[/perl]