在R里面实现这个功能其实非常简单,难的是很多packages经常会出现安装问题,更有的人压根不看芯片平台是什么,芯片对应的package是什么,就开始到处发问,自学能力实在是堪忧!
我前面有写目前所有bioconductor支持的芯片平台对应关系:通过bioconductor包来获取所有的芯片探针与gene的对应关系
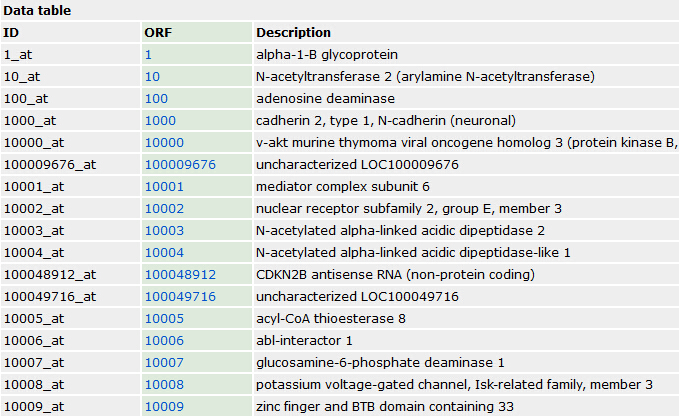
但那其实是一个很笨的办法,得到所有的各式各样的探针ID与基因的对应关系,以为它绕路了,正常情况只需要在GEO里面找到芯片对应基因关系即可,没必要下载那么多package的,但是这样做的好处也是很明显的, 对很多初学者来说,如果package能解决的话,就省心很多,比如下面这个转换关系:
suppressPackageStartupMessages(library(CLL))
## 这个package自带了一个数据,是我们需要用的
data(sCLLex) ## 这个数据里面有24个样本,分成两组,可以直接拿来测试差异基因分析
library(hgu95av2.db) ## 一定要搞清楚自己的芯片是什么数据包
exprSet=exprs(sCLLex) ##得到表达数据矩阵,但是矩阵的行名,是探针ID,无法理解,需要转换
##首先你取出所有的探针ID,#这里可以用三种方法来得到symbol,或者得到entrezID也可以
probeset=rownames(exprSet)
Symbol=as.character(as.list(hgu95av2SYMBOL[probeset]))
#annotate包提供 getSYMBOL( probeset ,"hgu95av2" )
#还可以用lookUp函数 lookUp( probeset , "hgu95av2", "SYMBOL")
#这些只是技巧而已啦
a=cbind.data.frame(Symbol,exprSet)
## 下面这个函数是对每个基因挑选最大表达量探针
rmDupID <-function(a=matrix(c(1,1:5,2,2:6,2,3:7),ncol=6)){
exprSet=a[,-1]
rowMeans=apply(exprSet,1,function(x) mean(as.numeric(x),na.rm=T))
a=a[order(rowMeans,decreasing=T),]
exprSet=a[!duplicated(a[,1]),]
#exprSet=apply(exprSet,2,as.numeric)
exprSet=exprSet[!
is.na(exprSet[,1]),]
rownames(exprSet)=exprSet[,1]
exprSet=exprSet[,-1]
return(exprSet)
}
exprSet=rmDupID(a)
对每个基因挑选最大表达量探针,只是一种处理方法而已,只是我一般处理芯片是这样做的,并不一定就是最好的!