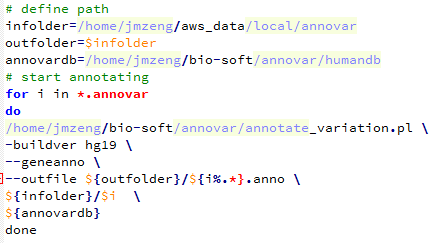
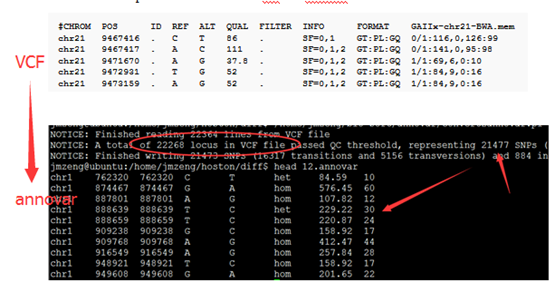
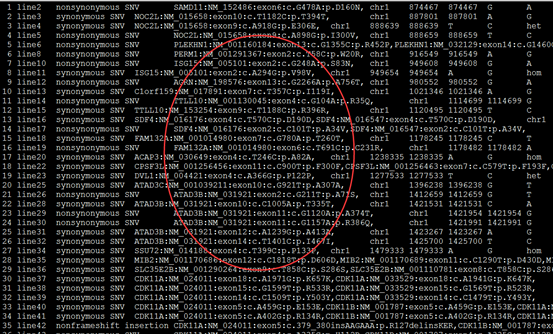
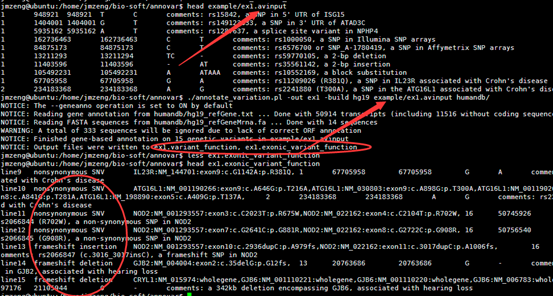
前面我已经讲了如何用annovar来把vcf格式的snp进行注释,注释之后大概是这样的,每个snp位点的坐标,已经在哪个基因上面,都标的很清楚啦,。而且该突变是在哪个基因的哪个转录本的哪个外显子都一清二楚,更强大的是,还能显示是第几个碱基突变成第几个,同样氨基酸的突变情况也很清楚。
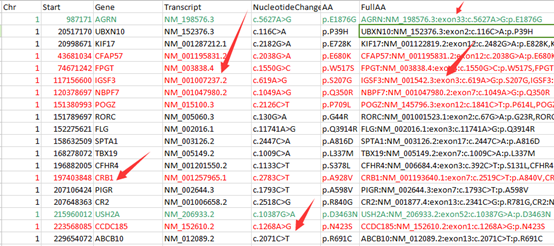
但是这样不是很方便浏览具体突变情况,所以我写了一个脚本格式化该突变情况。
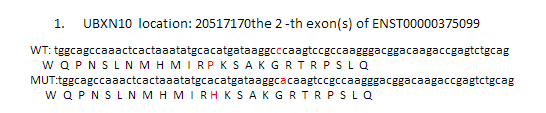
理论上是应该要做出上面这个样子,突变氨基酸前后各12个氨基酸都显示出来,突变的那个还要标红色突出显示!但是颜色控制很麻烦,我就没有做。效果如下
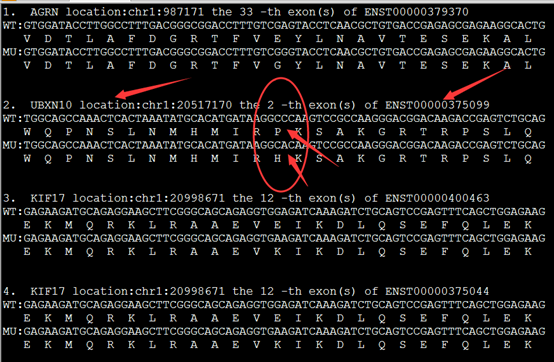
实现这样的格式化输出有三个重点,首先是NM开头的refseq的ID号要转换为ensembl数据库的转录本ID号,还有找到该转录本的CDS序列,这个都需要在biomart里面转换,或者自己写脚本,然后就用脚本爬取即可!
代码如下
[perl]
open FH1,"NM2ensembl.txt";
while(<FH1>){
chomp;
@F=split;
$hash_nm_enst{$F[4]}=$F[1] if $F[4];
}
open FH2,"ENST.CDS.fa";
while($line=<FH2>){
chomp $line;
if ($line=~/>/) {$key = (split /\|/,$line)[1];}
else {$hash_nucl{$key}.=$line;}
}
open FH3,"ENST.protein";
while($line=<FH3>){
chomp $line;
if ($line=~/>/) {$key = (split /\|/,$line)[1];}
else {$hash_prot{$key}.=$line;}
}
open FH4,"raw.mutiple.txt";
$i=1;
while(<FH4>){
chomp;
@F=split;
@tmp=split/:/,$F[1];
/:exon(\d+):/;$exon=$1;
/(NM_\d+)/; $nm=$1;
$enst=$hash_nm_enst{$nm};
print "$i. $tmp[0] $F[0] the $exon -th exon(s) of $enst \n";
$i++;
$tmp[3]=~/(\d+)/;$num_nucl=$1;
$tmp[3]=~/>([ATCG])/;$mutation_nucl=$1;
$tmp[4]=~/(\d+)/;$num_prot=$1;
$sequence=$hash_nucl{$enst};
$num_up=3*$num_prot-39;
$out_nucl=substr($sequence,$num_up,75);
print "WT:$out_nucl\n ";
for(my $j=0; $j < (length($out_nucl) - 2) ; $j += 3)
{print ' ';print $codon{substr($out_nucl,$j,3)} ;print ' ';}
print "\n";
$mutation_pos=$num_nucl-$num_up-1;
substr($out_nucl,$mutation_pos,1,$mutation_nucl) if ((length $out_nucl) == 75 );
print "MU:$out_nucl\n ";
for(my $j=0; $j < (length($out_nucl) - 2) ; $j += 3)
{print ' ';print $codon{substr($out_nucl,$j,3)} ;print ' ';}
print "\n";
print "\n";
print "\n";
}
[/perl]