BayesPeak也是peaks caller家族一员,用的人也不少,我这次也试了一下,因为是R的bioconductor系列包,所以直接在R里面安装就好,但是有几个点需要注意,我比对的基因组不只是Chr1~22,X,Y,M,还有一些contig和scaffold,需要在bam文件里面去除的,而且BayesPeak比较支持读取BED文件,可以直接转为GRanges对象,虽然它号称可以使用多核,但是计算速度还是非常慢。 Continue reading
七
05
BayesPeak也是peaks caller家族一员,用的人也不少,我这次也试了一下,因为是R的bioconductor系列包,所以直接在R里面安装就好,但是有几个点需要注意,我比对的基因组不只是Chr1~22,X,Y,M,还有一些contig和scaffold,需要在bam文件里面去除的,而且BayesPeak比较支持读取BED文件,可以直接转为GRanges对象,虽然它号称可以使用多核,但是计算速度还是非常慢。 Continue reading
CHIP-seq测序的本质还是目标片段捕获测序,跟WES不同的是,它不是通过固定的芯片探针来固定的捕获基因组上面特定序列,而是根据你选择的IP不同,你细胞或者机体状态不同,捕获到的序列差异很大!而我们研究的重点,就是捕获到的差异。而我们对CHIP-seq测序数据寻找peaks的本质就是得到所有测序数据比对在全基因组之后在正个基因组上面的测序深度里面寻找比较突出的。比如对WES数据来说,各个外显子,或者外显子的5端到3端,理论上测序深度应该是一致的,都是50X~200X,画一个测序深度曲线,应该是近似于一条直线。对我们的CHIP-seq测序数据来说,在所捕获的区域上面,理论上测序深度是绝对不一样的,应该是近似于一个山峰。而那些覆盖度高的地方,山顶,就是我们的IP所结合的热点,也就是我们想要找的peaks,在IGV里面看到大致是下面这样:
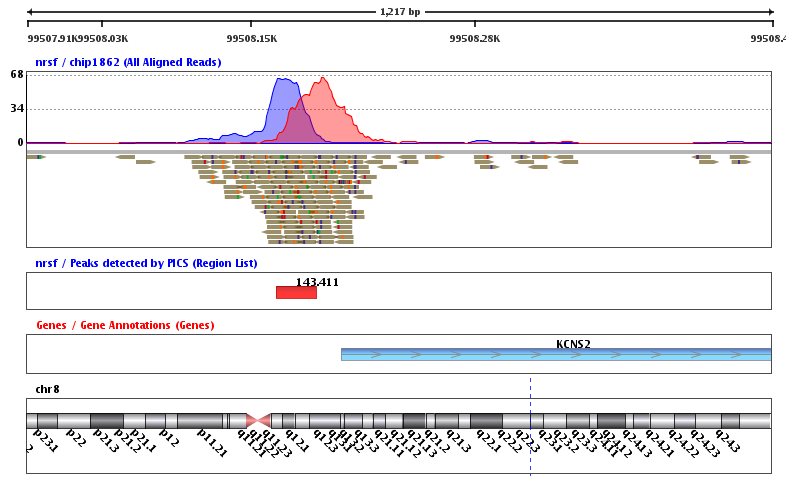
可以看到测序的reads分布是绝对的不均匀的!我们通常说的CHIP-seq测序的IP,可以是各个组蛋白的各个修饰位点对应的抗体,或者是各种转录因子的抗体,等等
如何定义热点呢?通俗地讲,热点是这样一些位置,这些位置多次被测得的read所覆盖(我们测的是一个细胞群体,read出现次数多,说明该位置被TF结合的几率大)。那么,read数达到多少才叫多?这就要用到统计检验喽。假设TF在基因组上的分布是没有任何规律的,那么,测序得到的read在基因组上的分布也必然是随机的,某个碱基上覆盖的read的数目应该服从二项分布。
具体统计学原理直接看原创吧:http://www.plob.org/2014/05/08/7227.html