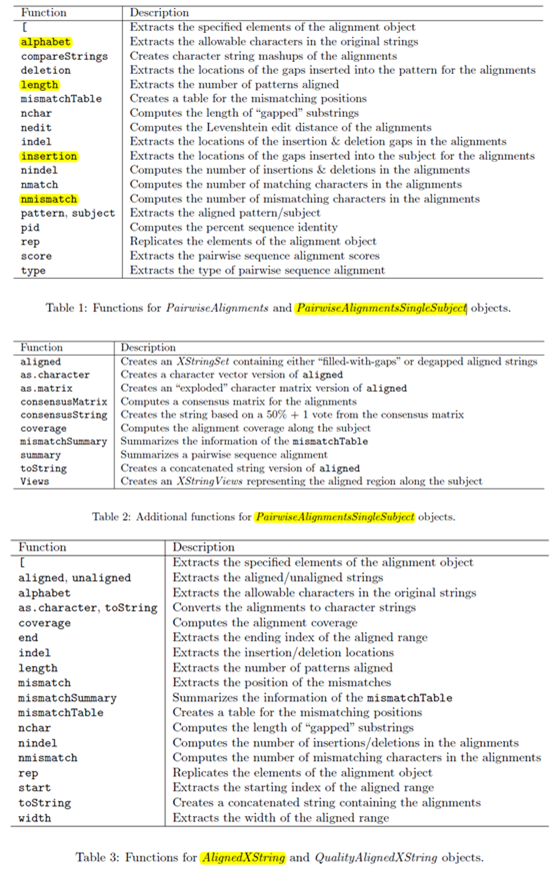
首先讲讲它的对象
有下面几个字符串对象BString, DNAString, RNAString and AAString可以通过以下代码构造它们:
b <- BString("I am a BString object")
d <- DNAString("TTGAAAA-CTC-N")
这两个对象的区别是DNAstring对象对字符串的要求严格一些,只有IUPAC字符和+-字符可以。
对构造好的对象可以通过下标来取子字符串对象,也可以通过subseq来取,但是子字符串仍然是数据对象,只有通过toString函数才能把它们转化成字符串。
用length(dd2)和nchar(toString(dd2))都可以找到我们Biostrings对象的长度。但是后者速度会很慢。
Views(RNAString("AU"), start=0, end=2)这个函数可以把string对象任意截取成list
start, end and width可以作用于我们截取的list,判断list里面的元素在原来的string对象上面的起始终止及长度信息。
接下来讲这个包带有的一个比对函数!
> pairwiseAlignment(pattern = c("succeed", "precede"), subject = "supersede")
Global PairwiseAlignmentsSingleSubject (1 of 2)pattern: [1] succ--eed subject: [1] supersede score: -33.99738
> pairwiseAlignment(pattern = c("succeed", "precede"), subject = "supersede",type = "local")
Local PairwiseAlignmentsSingleSubject (1 of 2)pattern: [1] su subject: [1] su score: 5.578203
> pairwiseAlignment(pattern = c("succeed", "precede"), subject = "supersede",gapOpening = 0, gapExtension = 1)
Global PairwiseAlignmentsSingleSubject (1 of 2)pattern: [1] su-cce--ed subject: [1] sup--
可以看出这个比对函数可以调整的参数实在是太多了,而且改变参数之后比对情况大不一样,还有很多参数就不一一细讲了。
这个比对结果可以赋值给一个变量,保存比对的对象。
psa1 <- pairwiseAlignment(pattern = c("succeed", "precede"), subject = "supersede")
class(psa1)
summary(psa1)
class(pattern(psa1))
class(summary(psa1))
score(psa2)
还可以自己构建打分矩阵来进行比对。
submat <-
+ matrix(-1, nrow = 26, ncol = 26, dimnames = list(letters, letters))
diag(submat) <- 0
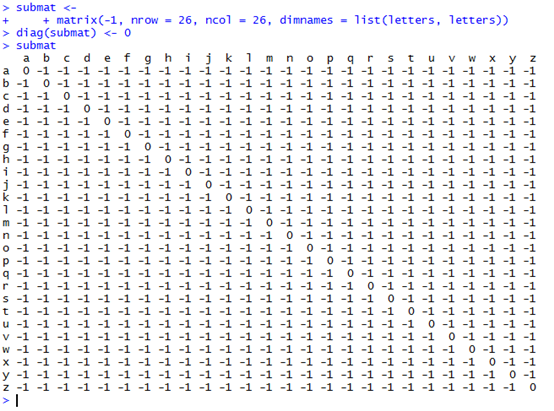
psa2 <-pairwiseAlignment(pattern = c("succeed", "precede"), subject = "supersede",substitutionMatrix = submat, gapOpening = 0, gapExtension = 1)
我们的包还自带了两个非常流行的氨基酸比对矩阵PAM和BLOSUM
ls("package:Biostrings")可以查看这个包所有的对象。
data(package="Biostrings")可以查看这个包所有的数据对象
还有很多其它函数
还可以去除adaptor,挺好玩的
既然有配对比对函数,那么就有多重比对函数!
我们可以读取clustaW, Phylip and Stolkholm这几种不同的比对结果文件来构造多重比对对象。
library(Biostrings)这个包里面自带了两个文件,我们可以示范一下构建对象。
origMAlign <- readDNAMultipleAlignment(filepath = system.file("extdata", "msx2_mRNA.aln", package="Biostrings"), format="clustal")
phylipMAlign <- readAAMultipleAlignment(filepath = system.file("extdata","Phylip.txt", package="Biostrings"),format="phylip")
对构造好的多重比对对象就可以构建进化树啦,代码如下!
sdist <- stringDist(as(origMAlign,"DNAStringSet"), method="hamming")
> clust <- hclust(sdist, method = "single")
> pdf(file="badTree.pdf")
> plot(clust)
> dev.off()