对于CHIP-seq数据处理完全是自学的,所以有很多细节得慢慢学习回来,这次记录的就是当我们把测序仪的fastq数据比对到参考基因组之后,应该对比对的结果文件做什么样的处理,然后去给peaks caller软件拿来call peaks呢?我看过博客 提到只保留比对质量值大于30的,也看过博客提到只保留unique比对的reads,我这里拿一篇公共数据测试了一下它们的区别!数据描述如下: Continue reading
八
07
对于CHIP-seq数据处理完全是自学的,所以有很多细节得慢慢学习回来,这次记录的就是当我们把测序仪的fastq数据比对到参考基因组之后,应该对比对的结果文件做什么样的处理,然后去给peaks caller软件拿来call peaks呢?我看过博客 提到只保留比对质量值大于30的,也看过博客提到只保留unique比对的reads,我这里拿一篇公共数据测试了一下它们的区别!数据描述如下: Continue reading
经过前面的CHIP-seq测序数据处理的常规分析,我们已经成功的把测序仪下机数据变成了BED格式的peaks记录文件,我选取的这篇文章里面做了4次CHIP-seq实验,分别是两个重复的野生型MCF7细胞系的 BAF155 immunoprecipitates和两个重复的突变型MCF7细胞系的 BAF155 immunoprecipitates,这样通过比较野生型和突变型MCF7细胞系的 BAF155 immunoprecipitates的结果的不同就知道该细胞系的BAF155 突变,对它在全基因组的结合功能的影响啦。
#我这里直接从GEO里面下载了peaks结果,它们详情如下:wc -l *bed
6768 GSM1278641_Xu_MUT_rep1_BAF155_MUT.peaks.bed
3660 GSM1278643_Xu_MUT_rep2_BAF155_MUT.peaks.bed
11022 GSM1278645_Xu_WT_rep1_BAF155.peaks.bed
5260 GSM1278647_Xu_WT_rep2_BAF155.peaks.bed
49458 GSM601398_Ini1HeLa-peaks.bed
24477 GSM601398_Ini1HeLa-peaks-stringent.bed
12725 GSM601399_Brg1HeLa-peaks.bed
12316 GSM601399_Brg1HeLa-peaks-stringent.bed
46412 GSM601400_BAF155HeLa-peaks.bed
37920 GSM601400_BAF155HeLa-peaks-stringent.bed
30136 GSM601401_BAF170HeLa-peaks.bed
25432 GSM601401_BAF170HeLa-peaks-stringent.bed
每个BED的peaks记录,本质是就3列是需要我们注意的,就是染色体,以及在该染色体上面的起始和终止坐标,如下:
#PeakID chr start end strand Normalized Tag Count region size findPeaks Score Clonal Fold Change
chr20 52221388 52856380 chr20-8088 41141 +
chr20 45796362 46384917 chr20-5152 31612 +
chr17 59287502 59741943 chr17-2332 29994 +
chr17 59755459 59989069 chr17-667 19943 +
chr20 52993293 53369574 chr20-7059 12642 +
chr1 121482722 121485861 chr1-995 9070 +
chr20 55675229 55855175 chr20-6524 7592 +
chr3 64531319 64762040 chr3-4022 7213 +
chr20 49286444 49384563 chr20-4482 6165 +
我们所谓的peaks注释,就是想看看该peaks在基因组的哪一个区段,看看它们在各种基因组区域(基因上下游,5,3端UTR,启动子,内含子,外显子,基因间区域,microRNA区域)分布情况,但是一般的peaks都有近万个,所以需要批量注释,如果脚本学的好,自己下载参考基因组的GFF注释文件,完全可以自己写一个,我这里会介绍一个R的bioconductor包ChIPpeakAnno来做CHIP-seq的peaks注释,下面的包自带的示例:
library(ChIPpeakAnno)
bed <- system.file("extdata", "MACS_output.bed", package="ChIPpeakAnno")
gr1 <- toGRanges(bed, format="BED", header=FALSE)
## one can also try import from rtracklayer
library(rtracklayer)
gr1.import <- import(bed, format="BED")
identical(start(gr1), start(gr1.import))
gr1[1:2]
gr1.import[1:2] #note the name slot is different from gr1
gff <- system.file("extdata", "GFF_peaks.gff", package="ChIPpeakAnno")
gr2 <- toGRanges(gff, format="GFF", header=FALSE, skip=3)
ol <- findOverlapsOfPeaks(gr1, gr2)
makeVennDiagram(ol)##还可以用binOverFeature来根据特定的GRanges对象(通常是TSS)来画分布图
## Distribution of aggregated peak scores or peak numbers around transcript start sites.
可以看到这个包使用起来非常简单,只需要把我们做好的peaks文件(GSM1278641_Xu_MUT_rep1_BAF155_MUT.peaks.bed等等)用toGRanges或者import读进去,成一个GRanges对象即可,上面的代码是比较两个peaks文件的overlap。然后还可以根据R很多包都自带的数据来注释基因组特征:
data(TSS.human.GRCh37) ## 主要是借助于这个GRanges对象来做注释,也可以用getAnnotation来获取其它GRanges对象来做注释
## featureType : TSS, miRNA, Exon, 5'UTR, 3'UTR, transcript or Exon plus UTR
peaks=MUT_rep1_peaks
macs.anno <- annotatePeakInBatch(peaks, AnnotationData=TSS.human.GRCh37,
output="overlapping", maxgap=5000L)## 得到的macs.anno对象就是已经注释好了的,每个peaks是否在基因上,或者距离基因多远,都是写的清清楚楚
if(require(TxDb.Hsapiens.UCSC.hg19.knownGene)){
aCR<-assignChromosomeRegion(peaks, nucleotideLevel=FALSE,
precedence=c("Promoters", "immediateDownstream",
"fiveUTRs", "threeUTRs",
"Exons", "Introns"),
TxDb=TxDb.Hsapiens.UCSC.hg19.knownGene)
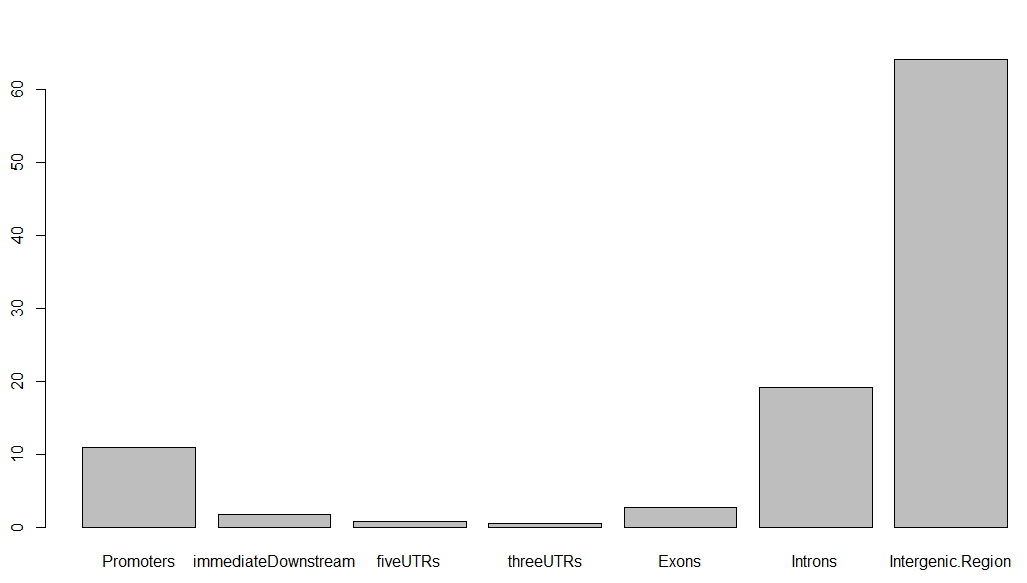
barplot(aCR$percentage)
}
得到的条形图如下,虽然很丑,但这就是peaks注释的精髓,搞清楚每个peaks在基因组的位置特征:
同理,对每个peaks文件,都可以做类似的分析!
但是对多个peaks文件,比如本文中的,想比较野生型和突变型MCF7细胞系的 BAF155 immunoprecipitates的结果的不同,就需要做peaks之间的差异分析,已经后续的差异基因注释啦
当然,值得一提的是peaks注释我更喜欢网页版工具,反正peaks文件非常小,直接上传到别人做好的web tools,就可立即出一大堆可视化图表分析结果啦,大家可以去试试看: