这个包主要有bam文件测试数据
> biocLite("pasillaBamSubset")
BioC_mirror: http://bioconductor.orgUsing Bioconductor version 3.0 (BiocInstaller 1.16.5), R version 3.1.2.
Installing package(s) 'pasillaBamSubset'
trying URL 'http://bioconductor.org/packages/3.0/data/experiment/bin/windows/contrib/3.1/pasillaBamSubset_0.3.1.zip'
Content type 'application/zip' length 31514402 bytes (30.1 Mb)
打开pasillaBamSubset包的安装地址就可以看到里面有几个bam文件
Several functions are available for reading BAM files into R:
而且加载包的同时也引入了几个读取bam文件的函数
readGAlignments()
readGAlignmentPairs()
readGAlignmentsList()
scanBam()
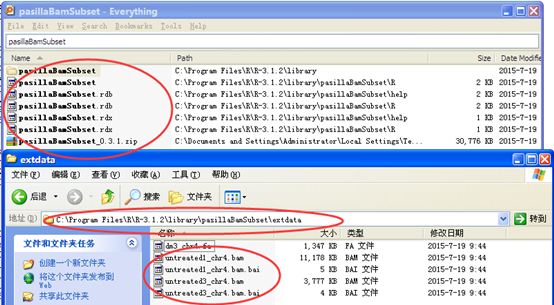
加载包就可以看到用两个函数得到包自带的数据文件的地址,主要是有很多人不一定把包安装在C盘,所以用函数来定位文件更加安全一点
> library(pasillaBamSubset)
> untreated1_chr4()
[1] "C:/Program Files/R/R-3.1.2/library/pasillaBamSubset/extdata/untreated1_chr4.bam"
> untreated3_chr4()
[1] "C:/Program Files/R/R-3.1.2/library/pasillaBamSubset/extdata/untreated3_chr4.bam"
接下来我们就看看如何读取这些bam文件的
library(pasillaBamSubset)
un1 <- untreated1_chr4() # single-end reads
library(GenomicAlignments)
reads1 <- readGAlignments(un1)
cvg1 <- coverage(reads1)
查看reads1这个结果,可以看到把这个bam文件都读成了一个数据对象GAlignments object,
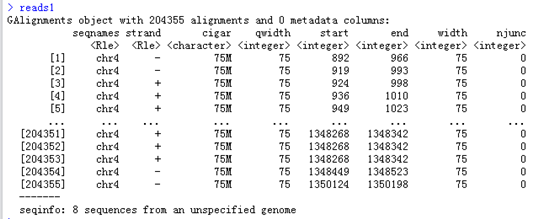
针对着个数据对象有很多操作,其中一个coverage操作是来自于GenomicFeatures
或者GenomicAlignments函数的,可以算出测序覆盖情况。
可以看到这个bam文件里面的比对情况大多几种在4号染色体里面
> cvg1$chr4
integer-Rle of length 1351857 with 122061 runs
Lengths: 891 27 5 12 13 45 5 12 13 ... 5 1 1 3 10
Values : 0 1 2 3 4 5 4 3 2 ... 12 11 10 6
> mean(cvg1$chr4)
[1] 11.33746
> max(cvg1$chr4)[1] 5627
可以看到平均测序深度是11.3X,最大测序深度是5627X