博文的顺序有点乱,因为怕读到前面的公共测序数据下载这篇文章的朋友搞不清楚,我如何调用各种软件的,所以我这里强势插入一篇博客来描述这件事,当然也只是略过,我所有的软件理论上都是安装在我的home目录下的biosoft文件夹,所以你看到我一般安装程序都是:
cd ~/biosoft
mkdir macs2 && cd macs2 ##指定的软件安装在指定文件夹里面 Continue reading
博文的顺序有点乱,因为怕读到前面的公共测序数据下载这篇文章的朋友搞不清楚,我如何调用各种软件的,所以我这里强势插入一篇博客来描述这件事,当然也只是略过,我所有的软件理论上都是安装在我的home目录下的biosoft文件夹,所以你看到我一般安装程序都是:
cd ~/biosoft
mkdir macs2 && cd macs2 ##指定的软件安装在指定文件夹里面 Continue reading
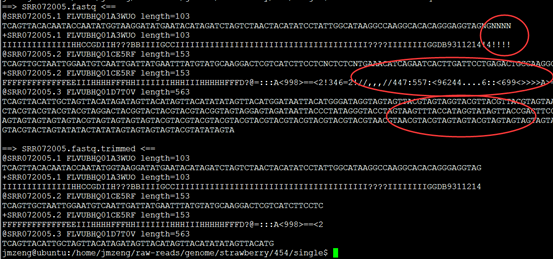
今天先 对7个单端数据做处理,是454数据,平均长度300bp左右,明天再处理3KB和20KB的配对reads。
首先跑fastqc
打开一个个看结果
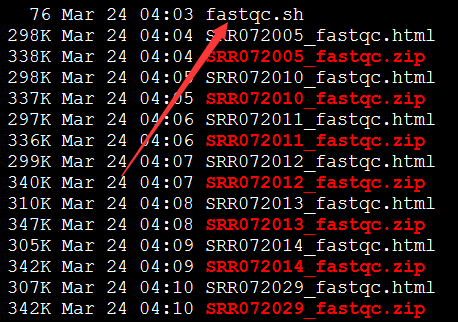
可以看到前面一些碱基的质量还是不错的, 因为这是454平台测序数据,序列片段长度差异很大,一般前四百个bp的碱基质量还是不错的,太长了的测序片段也不可靠
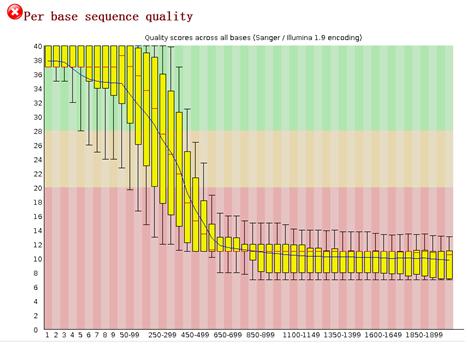
重点在下面这个图片,可以看到,前面的4个碱基是adaptor,肯定是要去除的,不是我们的测序数据。是TCAG,需要去除掉。
所以我们用了 solexaQA 这个套装软件对原始测序数据进行过滤
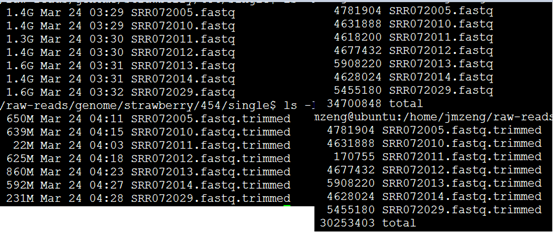
可以看到过滤的非常明显!!!甚至有个样本基本全军覆没了!然后我查看了我的批处理脚本,发现可能是perl DynamicTrim.pl -454 $id这个参数有问题
for id in *fastq
do
echo $id
perl DynamicTrim.pl -454 $id
done
for id in *trimmed
do
echo $id
perl LengthSort.pl $id
done
可以看到末尾的质量差的碱基都被去掉了,但是头部的TCAG还是没有去掉。
处理完毕后的数据如下:
仿写fastqc软件的一些功能(下)
文件来自于上面perl代码的输出文件,好像算法有点问题,26G的文件居然处理近一个小时才出数据!
R语言本身自带的画图工具都很丑,懒得说了,可以用ggplot2来重新画一个,不是项目要求没有报酬我就懒得画了,大家面前看看画图原理即可。
仿写fastqc软件的部分功能(上)
前面我们介绍了fastqc这个软件的使用方法 http://www.bio-info-trainee.com/?p=95 ,这是一个java软件,但是有些人服务器没有配置好这个java环境,导致无法使用,这里我贴出几个perl代码,也能实现fastqc的部分功能
统一测试文件是illumina的phred33格式的fastq文件,共100000/4=25000条reads,读长都是101个碱基
程序名-fastq2quality.pl
使用命令:perl fastq2quality.pl SRR504517_1.fastq >quality.txt
功能: 把fastq格式的每条原始reads的第四行ascii码质量值,转换为Q值并输出一个矩阵,有多少条reads就有多少行,每条reads的碱基数就是列数。