如果你研究癌症,那么TCGA计划的如此丰富的公共数据你肯定不能错过,一般人只能获取到level3的数据,当然,其实一般人也没办法使用level1和level2的数据,毕竟近万个癌症样本的原始测序数据,还是很恐怖的,而且我们拿到原始数据,再重新跑pipeline,其实并不一定比人家TCGA本身分析的要好,所以我们直接拿到分析结果,就足够啦!
五
06
如果你研究癌症,那么TCGA计划的如此丰富的公共数据你肯定不能错过,一般人只能获取到level3的数据,当然,其实一般人也没办法使用level1和level2的数据,毕竟近万个癌症样本的原始测序数据,还是很恐怖的,而且我们拿到原始数据,再重新跑pipeline,其实并不一定比人家TCGA本身分析的要好,所以我们直接拿到分析结果,就足够啦!
mutation signature这个概念提出来还不久,我看了看文献,最早见于2013年的一篇nature文章,主要是用来描述癌症患者的somatic mutation情况的。
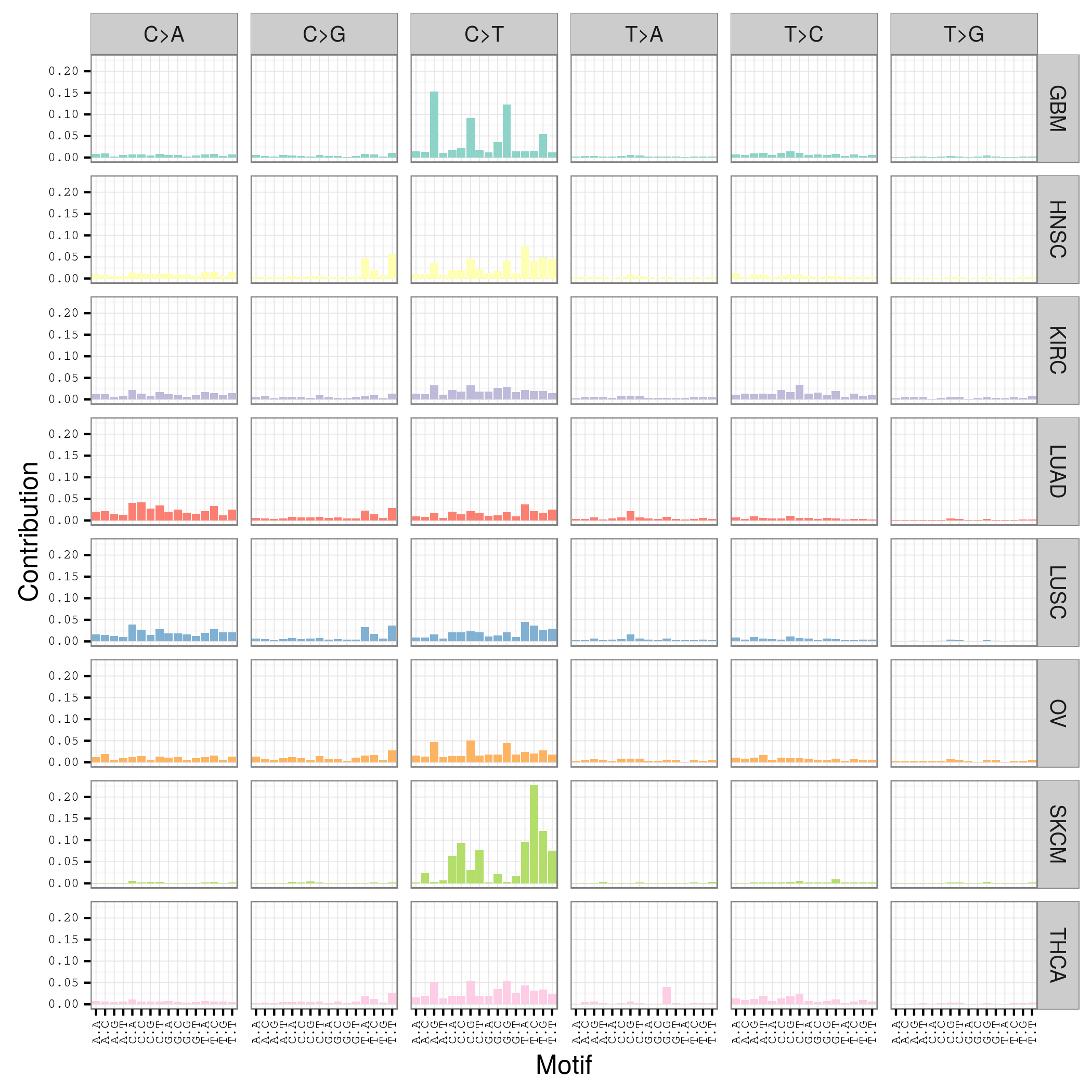
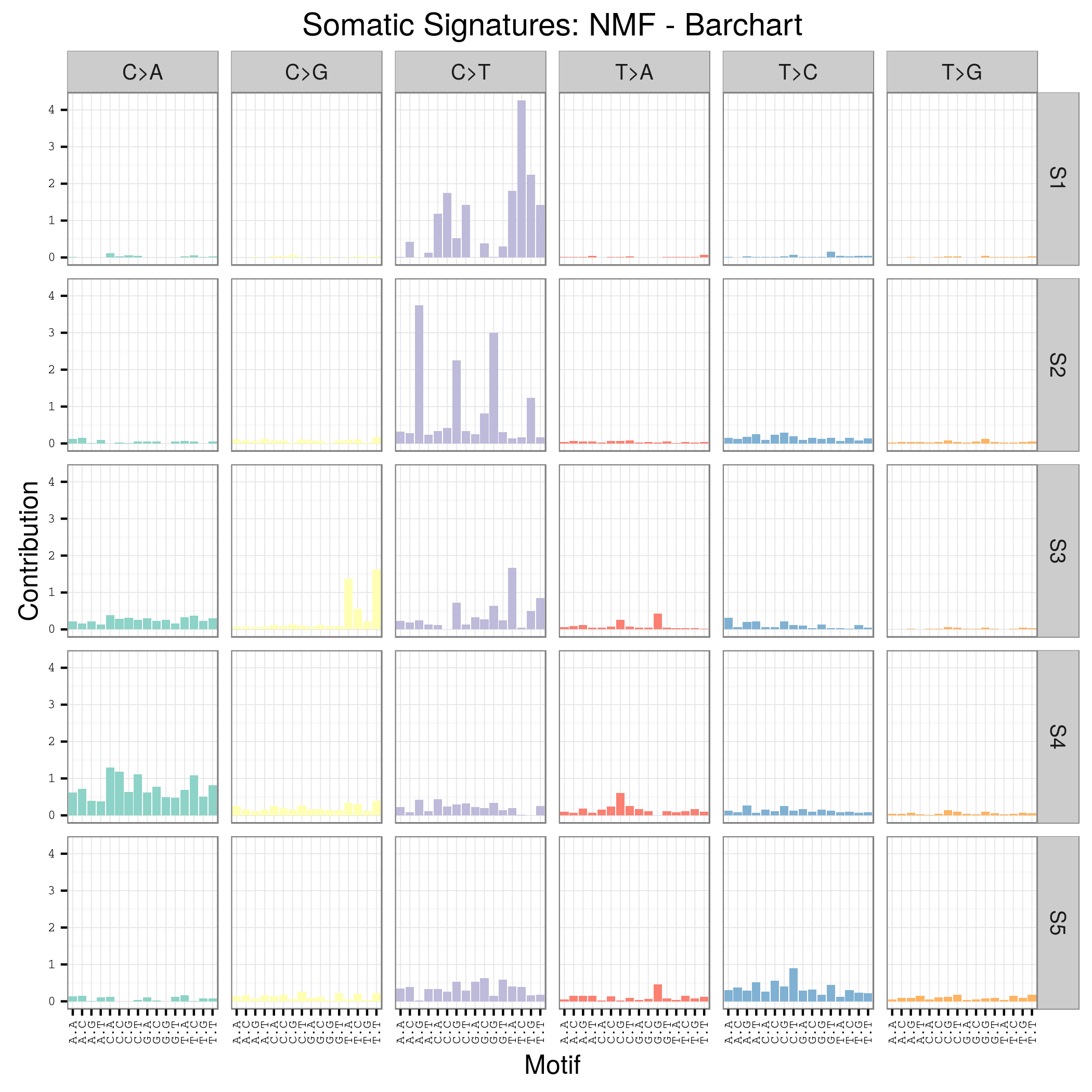
首先要自己分析癌症样本数据,拿到somatic mutation,TCGA计划发展到现在已经有非常多的somatic mutation结果啦,大家可以自行选择感兴趣的癌症数据拿来研究,解析一下mutation signature 。
我这里给大家推荐一个工具,是R语言的Bioconductor系列包中的一个,SomaticSignatures
其实它的说明书写的非常详细了已经,如果你理解了mutation signature的概念,很容易用那个包,其实你自己写一个脚本也是非常任意的,就是根据mutation的位置在基因组中找到它的前后一个碱基,然后组成三碱基突变模式,最后统计一下那96种突变模式的分布状况!
我这里简单讲一讲这个包如何用吧!
首先下载并加载几个必须的包:
然后根据突变数据做好一个GRanges对象,这个可以看我以前的博客
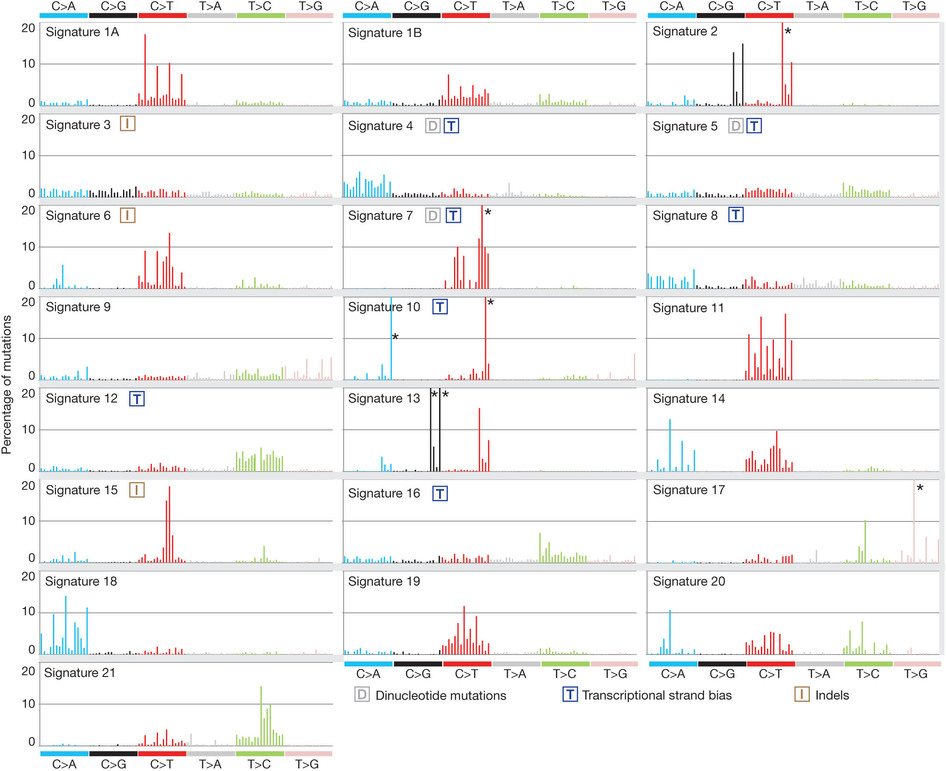
这也是对TCGA数据的深度挖掘,从而提出的一个统计学概念。文章研究了30种癌症,发现21种不同的mutation signature。如果理解了,就会发现这个其实蛮简单的,他们并不重新测序,只是拿已经有了的TCGA数据进行分析,而且居然是发表在nature上面!
研究了4,938,362 mutations from 7,042 cancers样本,突变频谱的概念只是针对于somatic 的mutation。一般是对癌症病人的肿瘤组织和癌旁组织配对测序,过滤得到的somatic mutation,一般一个样本也就几百个somatic 的mutation。
paper链接是:http://www.nature.com/nature/journal/v500/n7463/full/nature12477.html
来自于:https://www.biostars.org/p/19104/
Here are a few more, a summary of the other answers, and updated links:
For a much more general discussion of variant calling (not necessarily somatic or limited to SNVs/InDels) check out this thread: What Methods Do You Use For In/Del/Snp Calling?
Some papers describing comparisons of these callers:
The ICGC-TCGA DREAM Mutation Calling challenge has a component on somatic SNV calling.
This paper used validation data to compare popular somatic SNV callers:
Detecting somatic point mutations in cancer genome sequencing data: a comparison of mutation callers
You'll need to update the link to MuTect. Broad Institute has begun to put portable versions of their tools on Github, like thelatest release of MuTect. The Genome Institute at WashU has been using Github for a while, but portable versions of their tools can be found here and here.
To rehash/expand on what Dan said, if you're sequencing normal tissue, you generally expect to see single-nucleotide variant sites fall into one of three bins: 0%, 50%, or 100%, depending on whether they're heterozygous or homozygous.
With tumors, you have to deal with a whole host of other factors:
These, and other factors, make calling somatic variants difficult and still an area that is being heavily researched. If someone tells you that somatic variant calling is a solved problem, they probably have never tried to call somatic variants.
Sounds like somatic / tumor variant calling is something that will be solved by improvements at the wet lab side ( single cell selection / amplification / sequencing ) . Rather than at the computational side.
Well, single cell has a role to play (and would have more of one if WGA wasn't so lossy), but realistically, you can't sequence billions of cells from a tumor individually. Bulk sequencing still is going to have a role for quite a while.
Hell germ line calling isn't even a solved problem. Still get lots of false positives (and false negatives). It just tends to work so well that it is hard to improve it much except by making it faster, less memory intensive, etc
Solved was the wrong word. I just meant improved. There is only so much you can do at the computational side. Wet lab also has its part to play.
A germline variant caller generally has a ploidy-based genotyping algorithm built in to part of the algorithm/pipeline. I believe, IIRC, the GATK UnifiedGenotyper for instance does both variant calling and then genotype calling. So to call a genotype for a variant it is expecting a certain number of reads to support the alternative allele. When working with somatic variants all of the assumptions about how many reads you expect with a variant at a position to distinguish between true and false positives are no longer valid. Except for fixed mutations throughout the tumor population only some proportion of cells will hold a somatic variation. You also typically have some contamination from normal non-cancerous cells. Add in complications from significant genomic instability with lots of copy number variations and such and you have a need for a major change in your model for calling variation while minimizing artifactual calls. So you have a host of other programs that have been developed specifically for looking at somatic variation in tumor samples.
一篇文献:
是qiagen公司发的
High-throughput sequencing is rapidly becoming common practice in clinical diagnosis and cancer research. Many algorithms have been developed for somatic single nucleotide variant (SNV) detection in matched tumor-normal DNA sequencing. Although numerous studies have compared the performance of various algorithms on exome data, there has not yet been a systematic evaluation using PCR-enriched amplicon data with a range of variant allele fractions. The recently developed gold standard variant set for the reference individual NA12878 by the NIST-led “Genome in a Bottle” Consortium (NIST-GIAB) provides a good resource to evaluate admixtures with various SNV fractions.
Using the NIST-GIAB gold standard, we compared the performance of five popular somatic SNV calling algorithms (GATK UnifiedGenotyper followed by simple subtraction, MuTect, Strelka, SomaticSniper and VarScan2) for matched tumor-normal amplicon and exome sequencing data.
Nevertheless, detecting somatic mutations is still challenging, especially for low-allelic-fraction variants caused by tumor heterogeneity, copy number alteration, and sample degradation
We used QIAGEN’s GeneRead DNAseq Comprehensive Cancer Gene Panel (CCP, Version 1) for enrichment and library construction in triplicate。
QIAGEN’s GeneRead DNAseq Comprehensive Cancer Gene Panel (Version 1) was used to amplify the target region of interest (124 genes, 800 Kb).
When analyzing different types of data, use of different algorithms may be appropriate.
DNA samples of NA12878 and NA19129 were purchased from Coriell Institute. Sample mixtures were created based on the actual amplifiable DNA in each sample, resulting in 0%, 8%, 16%, 36%, and 100% of NA12878 sample mixed in the NA19129 sample, respectively.We treated the mixed samples at 8%, 16%, 36%, and 100% as the virtual tumor samples and the 0% as the virtual normal sample.
五个软件的算法是:
1. NaiveSubtract — SNVs were called separately from virtual tumor and normal samples using GATK UnifiedGenotyper [22]. For exome sequencing data, reads were already mapped, locally realigned and recalibrated by the 1,000 Genomes Project. So SNVs were directly called on the BAM files using GATK Unified Genotyper. Then, SNVs detected in the virtual normal sample were removed from the list of SNVs detected in the virtual tumor sample, leaving the “somatic” SNVs.
2. MuTect — MuTect is a method developed for detecting the most likely somatic point mutations in NGS data using a Bayesian classifier approach. The method includes pre-processing aligned reads separately in tumor and normal samples and post-processing resulting variants by applying an additional set of filters. We ran MuTect under the High-Confidence mode with its default parameter settings. We disabled the “Clustered position” filter and the “dbSNP filter” for the amplicon sequencing reads, and we disabled the “dbSNP filter” for the exome sequencing.
3. SomaticSniper — SomaticSniper calculates the Bayesian posterior probability of each possible joint genotype across the normal and cancer samples. We tuned the software’s parameters to increase sensitivity and then filtered raw results using a Somatic Score cut-off of 20 to improve specificity.
4. Strelka — Strelka reports the most likely genotype for tumor and normal samples based on a Bayesian probability model. Post-calling filters built into the software are based on factors such as read depth, mismatches, and overlap with indels. We skipped depth filtration for exome and amplicon sequencing data as recommended by the Strelka authors. For the amplicon sequencing reads, we set the minimum MAPQ score at 17 for consistency with the defaults in GATK UnifiedGenotyper. We used variants passing Strelka post-calling filters for analysis.
5. VarScan2 — VarScan2 performs analyses independently on pileup files from the tumor and normal samples to heuristically call a genotype at positions achieving certain thresholds of coverage and quality. Then, sites of the genotypes not matched in tumor and normal samples are classified into somatic, germline, or ambiguous groups using Fisher’s exact test. We generated the pileup files using SAMtools mpileup command.
The compatibility of the output VCF files between different methods as well as the NIST-GIAB gold standard was examined using bcbio.variation tools and manual inspection. The reported SNP call representations between files are comparable to each other.
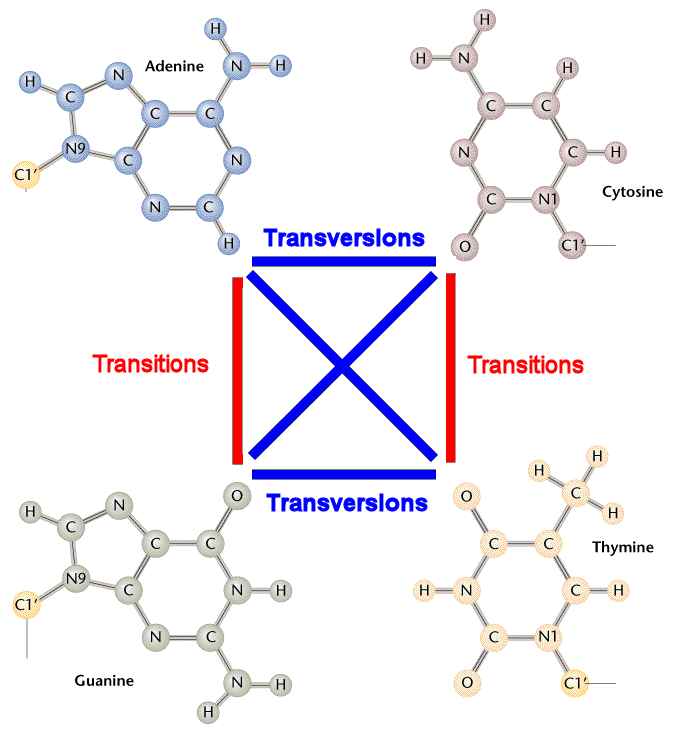
DNA分子中某一个碱基为另一种碱基置换,导致DNA碱基序列异常,是基因突变的一种类型。可分为转换和颠换两类。转换(transitions)是同类碱基的置换(AT→GC及GC→AT),颠换(transversions) 是不同类碱基的置换(AT→TA或CG,GC→CG或TA。
DNA substitution mutations are of two types. Transitions are interchanges of two-ring purines (A G) or of one-ring pyrimidines (C T): they therefore involve bases of similar shape. Transversions are interchanges of purine for pyrimidine bases, which therefore involve exchange of one-ring and two-ring structures.
我们在分析driver mutation的时候会区分各种点突变:
那么,我们有64种密码子,每种密码子都会有9种突变可能,我们如何得到一个所有的突变可能的分类并且打分表格呢?
类似于下面这样的表格:共576行!!!
head category.acgt
AAA>AAT 2 A T 6
AAA>AAC 2 A C 6
AAA>AAG 2 A G 5
AAA>ATA 1 A T 6
AAA>ACA 1 A C 6
AAA>AGA 1 A G 5
AAA>TAA 0 A T 6
AAA>CAA 0 A C 6
AAA>GAA 0 A G 5
AAT>AAA 2 T A 6
tail category.acgt
GGC>GGG 2 C G 2
GGG>AGG 0 G A 3
GGG>TGG 0 G T 4
GGG>CGG 0 G C 4
GGG>GAG 1 G A 3
GGG>GTG 1 G T 4
GGG>GCG 1 G C 4
GGG>GGA 2 G A 3
GGG>GGT 2 G T 4
GGG>GGC 2 G C 4
我本来以为这是一件很简单的事情,写起来,才发现好麻烦

里面用到的一个函数如下:就是判断突变属于上述六种的哪一种!

参考:https://www.mun.ca/biology/scarr/Transitions_vs_Transversions.html
https://en.wikipedia.org/wiki/Transversion
http://www.uvm.edu/~cgep/Education/Mutations.html
点突变,也称作单碱基替换(single base substitution),指由单个碱基改变发生的突变。
可以分为转换(transitions)和颠换(transversions)两类。
转换:嘌呤和嘌呤之间的替换,或嘧啶和嘧啶之间的替换。
颠换:嘌呤和嘧啶之间的替换。
方便理解下面再附上一张示意图,如下: