请不要直接拷贝我的代码,需要自己理解,然后打出来,思考我为什么这样写代码。
软件请用最新版,尤其是samtools等被我存储在系统环境变量的,考虑到读者众多,一般的软件我都会自带版本信息的!
我用两个小时,不代表你是两个小时就学会,有些朋友反映学了两个星期才 学会,这很正常,没毛病,不要异想天开两个小时就达到我的水平。
本次讲解选取的文章是为了探索PRC1,PCR2这样的蛋白复合物,不是转录因子或者组蛋白的CHIP-seq,请注意区别!
这是一个系列帖子,你可以先看:
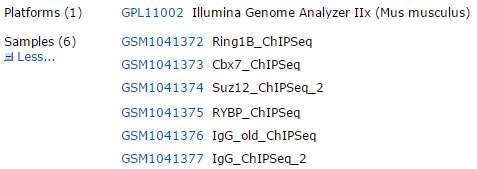
文章是:RYBP and Cbx7 define specific biological functions of polycomb complexes in mouse embryonic stem cells
RYBP and Cbx7都是Polycomb repressive complex 1 (PRC1)的组分:
所以用脚本在ftp里面批量下载即可: