这个软件不仅仅能做QC,而且可以统计各个基因的RPKM值!尤其是TCGA计划里面的都是用它算的
一、程序安装
但是需要下载很多注释数据

二、输入数据
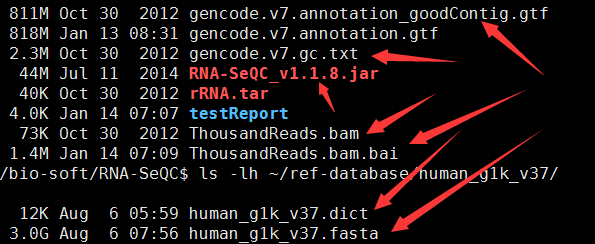
箭头所指的文件,一个都不少,只有那个rRNA.tar我没有用, 因为这个软件有两种使用方式,我用的是第一种
三、软件使用
软件的官网给力例子,很容易学习:
RNA-SeQC can be run with or without a BWA-based rRNA level estimation mode. To run without (less accurate, but faster) use the command:
java -jar RNASeQC.jar -n 1000 -s "TestId|ThousandReads.bam|TestDesc" -t gencode.v7.annotation_goodContig.gtf -r Homo_sapiens_assembly19.fasta -o ./testReport/ -strat gc -gc gencode.v7.gc.txt
我用的就是这个例子,这个例子需要的所有文件里面,染色体都是没有chr的,这个非常重要!!!
代码如下:
java -jar RNA-SeQC_v1.1.8.jar \
-n 1000 \
-s "TestId|ThousandReads.bam|TestDesc" \
-t gencode.v7.annotation_goodContig.gtf \
-r ~/ref-database/human_g1k_v37/human_g1k_v37.fasta \
-o ./testReport/ \
-strat gc \
-gc gencode.v7.gc.txt \
To run the more accurate but slower, BWA-based method :
java -jar RNASeQC.jar -n 1000 -s "TestId|ThousandReads.bam|TestDesc" -t gencode.v7.annotation_goodContig.gtf -r Homo_sapiens_assembly19.fasta -o ./testReport/ -strat gc -gc gencode.v7.gc.txt -BWArRNA human_all_rRNA.fasta
Note: this assumes BWA is in your PATH. If this is not the case, use the -bwa flag to specify the path to BWA
四、结果解读
运行要点时间,就那个一千条reads的测试数据都搞了10分钟!
出来一大堆突变,具体解释,官网上面很详细,不过,比较重要的当然是RPKM值咯,还有QC的信息
TCGA数据里面都会提供由RNA-SeQC软件处理得到的表达矩阵!
Expression
- RPKM data are used as produced by RNA-SeQC.
- Filter on >=10 individuals with >0.1 RPKM and raw read counts greater than 6.
- Quantile normalization was performed within each tissue to bring the expression profile of each sample onto the same scale.
- To protect from outliers, inverse quantile normalization was performed for each gene, mapping each set of expression values to a standard normal.
软件的主页是: