一、下载安装该软件。
网上可以搜索到下载地址,解压之后make即可
一般都会报错
In file included from bam_cat.c:41:0:
htslib-1.1/htslib/bgzf.h:34:18: fatal error: zlib.h: No such file or directory
#include <zlib.h>
^
compilation terminated.
make: *** [bam_cat.o] Error 1
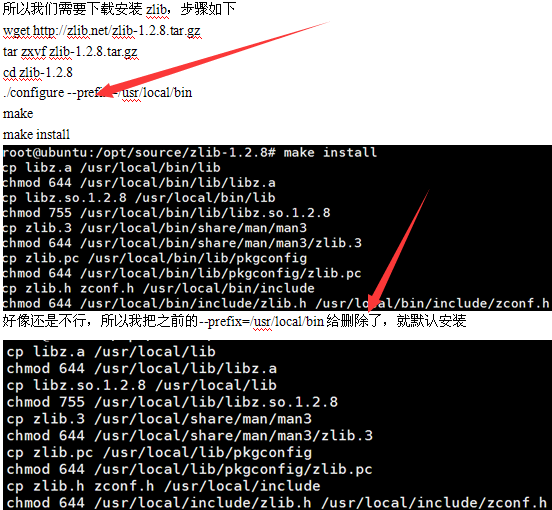
然后,居然就通过了,晕。有时候我实在是搞不定linux系统一些具体的原理,但是反正就是能用!学会搜索,学会试错即可。
直到两年后我才理解(linux下 的软件安装需要指定路径,而且是自己有权限的路径,2016年11月23日10:12:11),比如安装下面的方式来安装软件:
mkdir -p ~/biosoft/myBin
echo 'export PATH=/home/jianmingzeng/biosoft/myBin/bin:$PATH' >>~/.bashrc
source ~/.bashrc
cd ~/biosoft
mkdir cmake && cd cmake
wget http://cmake.org/files/v3.3/cmake-3.3.2.tar.gz
tar xvfz cmake-3.3.2.tar.gz
cd cmake-3.3.2
./configure --prefix=/home/jianmingzeng/biosoft/myBin ## 这里非常重要
make
make install
但是有些电脑会报另外一个错
#include <curses.h>
^
compilation terminated.
make: *** [bam_tview_curses.o] Error 1
我也顺便解决一下,因为以前我的服务器遇到过,也是很纠结的。
sudo apt-get install libncurses5-dev
二.准备数据及使用,见我的snp-caling流程
http://www.bio-info-trainee.com/?p=439
samtools view -bS tmp1.sam > tmp1.bam
samtools sort tmp1.bam tmp1.sorted
samtools index tmp1.sorted.bam
samtools mpileup -d 1000 -gSDf ../../../ref-database/hg19.fa tmp1.sorted.bam |bcftools view -cvNg – >tmp1.vcf
因为这个软件都是与bwa和bowtie等能产生sam文件的软件合作才能使用。
其中这个软件参数还是蛮多的,但是常用的就那么几个,网上也很容易找到教程
简单附上一点资料
samtools是一个用于操作sam和bam文件的工具合集。包含有许多命令。以下是常用命令的介绍
1. view
view命令的主要功能是:将sam文件转换成bam文件;然后对bam文件进行各种操作,比如数据的排序(不属于本命令的功能)和提取(这些操作是对bam文件进行的,因而当输入为sam文件的时候,不能进行该操作);最后将排序或提取得到的数据输出为bam或sam(默认的)格式。
bam文件优点:bam文件为二进制文件,占用的磁盘空间比sam文本文件小;利用bam二进制文件的运算速度快。
view命令中,对sam文件头部的输入(-t或-T)和输出(-h)是单独的一些参数来控制的。
Usage: samtools view [options] <in.bam>|<in.sam> [region1 [...]]默认情况下不加 region,则是输出所有的 region. Options:
-b output BAM 默认下输出是 SAM 格式文件,该参数设置输出 BAM 格式 -h print header for the SAM output 默认下输出的 sam 格式文件不带 header,该参数设定输出sam文件时带 header 信息 -H print header only (no alignments) -S input is SAM 默认下输入是 BAM 文件,若是输入是 SAM 文件,则最好加该参数,否则有时候会报错。
例子:
#将sam文件转换成bam文件$ samtools view -bS abc.sam > abc.bam$ samtools view -b -S abc.sam -o abc.bam
#提取比对到参考序列上的比对结果$ samtools view -bF 4 abc.bam > abc.F.bam #提取paired reads中两条reads都比对到参考序列上的比对结果,只需要把两个4+8的值12作为过滤参数即可$ samtools view -bF 12 abc.bam > abc.F12.bam #提取没有比对到参考序列上的比对结果$ samtools view -bf 4 abc.bam > abc.f.bam #提取bam文件中比对到caffold1上的比对结果,并保存到sam文件格式$ samtools view abc.bam scaffold1 > scaffold1.sam #提取scaffold1上能比对到30k到100k区域的比对结果$ samtools view abc.bam scaffold1:30000-100000 > scaffold1_30k-100k.sam #根据fasta文件,将 header 加入到 sam 或 bam 文件中$ samtools view -T genome.fasta -h scaffold1.sam > scaffold1.h.sam
2. sort
sort对bam文件进行排序。
Usage: samtools sort [-n] [-m <maxMem>] <in.bam> <out.prefix> -m 参数默认下是 500,000,000 即500M(不支持K,M,G等缩写)。对于处理大数据时,如果内存够用,则设置大点的值,以节约时间。-n 设定排序方式按short reads的ID排序。默认下是按序列在fasta文件中的顺序(即header)和序列从左往右的位点排序。
例子:
$ samtools sort abc.bam abc.sort$ samtools view abc.sort.bam | less -S
3.merge
将2个或2个以上的已经sort了的bam文件融合成一个bam文件。融合后的文件不需要则是已经sort过了的。
Usage: samtools merge [-nr] [-h inh.sam] <out.bam> <in1.bam> <in2.bam>[...] Options: -n sort by read names -r attach RG tag (inferred from file names) -u uncompressed BAM output -f overwrite the output BAM if exist -1 compress level 1 -R STR merge file in the specified region STR [all] -h FILE copy the header in FILE to <out.bam> [in1.bam] Note: Samtools' merge does not reconstruct the @RG dictionary in the header. Users must provide the correct header with -h, or uses Picard which properly maintains the header dictionary in merging.
4.index
必须对bam文件进行默认情况下的排序后,才能进行index。否则会报错。
建立索引后将产生后缀为.bai的文件,用于快速的随机处理。很多情况下需要有bai文件的存在,特别是显示序列比对情况下。比如samtool的tview命令就需要;gbrowse2显示reads的比对图形的时候也需要。
Usage: samtools index <in.bam> [out.index]
例子:
#以下两种命令结果一样$ samtools index abc.sort.bam$ samtools index abc.sort.bam abc.sort.bam.bai
5. faidx
对fasta文件建立索引,生成的索引文件以.fai后缀结尾。该命令也能依据索引文件快速提取fasta文件中的某一条(子)序列
Usage: samtools faidx <in.bam> [ [...]] 对基因组文件建立索引$ samtools faidx genome.fasta#生成了索引文件genome.fasta.fai,是一个文本文件,分成了5列。第一列是子序列的名称;第二列是子序列的长度;个人认为“第三列是序列所在的位置”,因为该数字从上往下逐渐变大,最后的数字是genome.fasta文件的大小;第4和5列不知是啥意思。于是通过此文件,可以定位子序列在fasta文件在磁盘上的存放位置,直接快速调出子序列。 #由于有索引文件,可以使用以下命令很快从基因组中提取到fasta格式的子序列$ samtools faidx genome.fasta scffold_10 > scaffold_10.fasta
6. tview
tview能直观的显示出reads比对基因组的情况,和基因组浏览器有点类似。
Usage: samtools tview <aln.bam> [ref.fasta] 当给出参考基因组的时候,会在第一排显示参考基因组的序列,否则,第一排全用N表示。按下 g ,则提示输入要到达基因组的某一个位点。例子“scaffold_10:1000"表示到达第10号scaffold的第1000个碱基位点处。使用H(左)J(上)K(下)L(右)移动显示界面。大写字母移动快,小写字母移动慢。使用空格建向左快速移动(和 L 类似),使用Backspace键向左快速移动(和 H 类似)。Ctrl+H 向左移动1kb碱基距离; Ctrl+L 向右移动1kb碱基距离可以用颜色标注比对质量,碱基质量,核苷酸等。30~40的碱基质量或比对质量使用白色表示;20~30黄色;10~20绿色;0~10蓝色。使用点号'.'切换显示碱基和点号;使用r切换显示read name等还有很多其它的使用说明,具体按 ? 键来查看。
参考:samtools的说明文档:http://samtools.sourceforge.net/samtools.shtml
http://www.plob.org/2014/01/26/7112.html