首先安装这个包
source("http://bioconductor.org/biocLite.R")
biocLite("seqinr")
然后加载包,并读取我们的CDS.fa文件
library("seqinr")
human_cds=read.fasta("CDS.fa")
#这一个步骤非常耗时间,可能是因为我们的转录本文件有十万多个转录本的原因吧
str(human_cds) #查看可知读入了一个list,其中每个转录本都是list的一个元素
List of 100778
$ ENST00000415118:Class 'SeqFastadna' atomic [1:8] g a a a ...
.. ..- attr(*, "name")= chr "ENST00000415118"
.. ..- attr(*, "Annot")= chr ">ENST00000415118 havana_ig_gene:known chromosome:GRCh38:14:22438547:22438554:1 gene:ENSG00000223997 gene_biotype:TR_D_gene tran"| __truncated__
$ ENST00000448914:Class 'SeqFastadna' atomic [1:13] a c t g ...
.. ..- attr(*, "name")= chr "ENST00000448914"
.. ..- attr(*, "Annot")= chr ">ENST00000448914 havana_ig_gene:known chromosome:GRCh38:14:22449113:22449125:1 gene:ENSG00000228985 gene_biotype:TR_D_gene tran"| __truncated__
对list的每个元素都有几种函数可以处理得到信息:
Length,table,GC,count
其中count函数很有趣,数一数序列里面的这些组合出现的次数
count(dengueseq, 1)
count(dengueseq, 2)接下来我们随机取human_cds这个list的一个元素用这几个函数对它处理一下
> tmp=human_cds[[1]]
> tmp
[1] "g" "a" "a" "a" "t" "a" "g" "t"
attr(,"name")
[1] "ENST00000415118"
attr(,"Annot")
[1] ">ENST00000415118 havana_ig_gene:known chromosome:GRCh38:14:22438547:22438554:1 gene:ENSG00000223997 gene_biotype:TR_D_gene transcript_biotype:TR_D_gene"
attr(,"class")
[1] "SeqFastadna"
再看看函数的结果
> length(tmp)
[1] 8
> table(tmp)
tmp
a g t
4 2 2
> GC(tmp)
[1] 0.25
> count(tmp,1)
a c g t
4 0 2 2
> count(tmp,2)
aa ac ag at ca cc cg ct ga gc gg gt ta tc tg tt
2 0 1 1 0 0 0 0 1 0 0 1 1 0 0 0
>
还是挺好用的,接下来我们应用R的知识来对着十万多个转录本进行一些简单的总结
human_cds_length=unlist(lapply(human_cds,length))
human_cds_gc=unlist(lapply(human_cds,GC))
这样就得到了所有转录本的长度和GC含量信息
然后我们简单统计一下,并画几个图表吧!
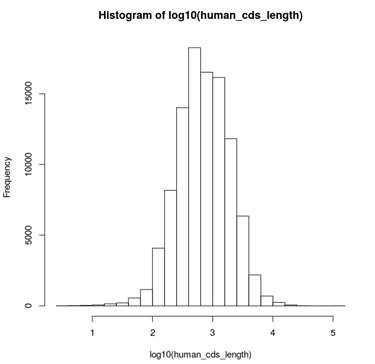
> summary(human_cds_length)
Min. 1st Qu. Median Mean 3rd Qu. Max.
3 366 699 1132 1425 108000
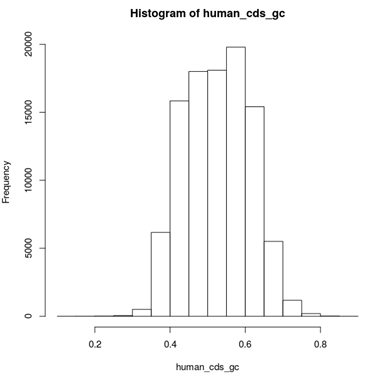
> summary(human_cds_gc)
Min. 1st Qu. Median Mean 3rd Qu. Max.
0.1467 0.4577 0.5285 0.5264 0.5932 0.8917
可以看到还是有很多很极端的转录本的存在!
最长的转录本也不过10k,而我记得最长的基因高达8M,看了内含子远大于外显子呀。
但是GC含量有很多高于80%,这些基因在二代测序的研究中是一个盲区。

这些极端基因可以通过biomaRt等包进行注释,得到gene名和功能信息。
hist(human_cds_gc)
hist(log10(human_cds_length))
GC含量分布如图
长度分布如图
附表:
http://www.bioinformatics.org/sms/iupac.html 所有字符的碱基氨基酸意义表格