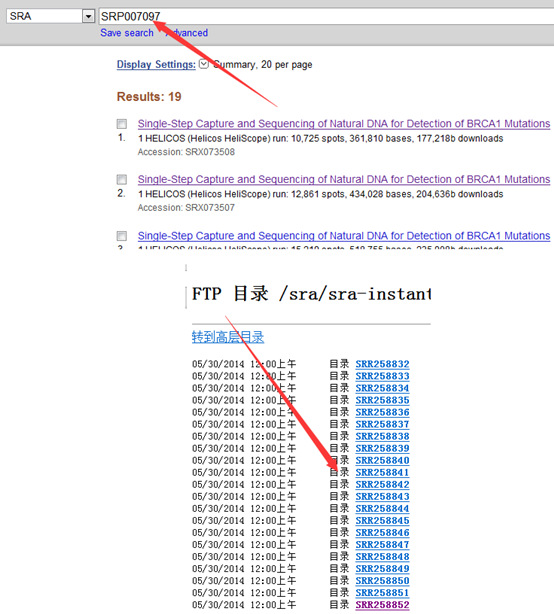
我这里拿的是bioconductor里面最常用的airway数据,因为差异表达分析在bioconductor里面是重点,它们这些包在介绍自己的算法以及做示范的时候都用的这个数据。可以在GEO数据库里面看到信息描述:http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE52778 可以看到是Illumina HiSeq 2000 (Homo sapiens) ,75bp paired-end 这个信息很重要,决定了下载sra数据之后如何解压以及如何比对。也可以看到作者把所有的测序原始数据都上传到了SRA中心:http://www.ncbi.nlm.nih.gov/sra?term=SRP033351 ,这里可以在linux服务器上面写一个简单的脚本批量下载所有的测序数据,然后根据GEO里面描述的metadata把原始数据改名。
for ((i=508;i<=523;i++)) ;do wget ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByStudy/sra/SRP/SRP033/SRP033351/SRR1039$i/SRR1039$i.sra;done
ls *sra |while read id; do ~/biosoft/sratoolkit/sratoolkit.2.6.3-centos_linux64/bin/fastq-dump --split-3 $id;done
需要自己看SRA里面的数据记录,上面的脚本不难写出,然后因为是Illumina的双端测序,所以我们用fastq-dump --split-3命令来把sra格式数据转换为fastq,但是因为这里有16个测序数据,所以最好是同步改名,我这里用脚本批量生成改名脚本如下:
为了节省空间,我用了--gzip压缩,该文件名,用-A参数。
nohup ~/biosoft/sratoolkit/sratoolkit.2.6.3-centos_linux64/bin/fastq-dump --split-3 --gzip -A N61311_untreated SRR1039508.sra &
nohup ~/biosoft/sratoolkit/sratoolkit.2.6.3-centos_linux64/bin/fastq-dump --split-3 --gzip -A N61311_Dex SRR1039509.sra &
nohup ~/biosoft/sratoolkit/sratoolkit.2.6.3-centos_linux64/bin/fastq-dump --split-3 --gzip -A N61311_Alb SRR1039510.sra &
nohup ~/biosoft/sratoolkit/sratoolkit.2.6.3-centos_linux64/bin/fastq-dump --split-3 --gzip -A N61311_Alb_Dex SRR1039511.sra &
nohup ~/biosoft/sratoolkit/sratoolkit.2.6.3-centos_linux64/bin/fastq-dump --split-3 --gzip -A N052611_untreated SRR1039512.sra &
nohup ~/biosoft/sratoolkit/sratoolkit.2.6.3-centos_linux64/bin/fastq-dump --split-3 --gzip -A N052611_Dex SRR1039513.sra &
nohup ~/biosoft/sratoolkit/sratoolkit.2.6.3-centos_linux64/bin/fastq-dump --split-3 --gzip -A N052611_Alb SRR1039514.sra &
nohup ~/biosoft/sratoolkit/sratoolkit.2.6.3-centos_linux64/bin/fastq-dump --split-3 --gzip -A N052611_Alb_Dex SRR1039515.sra &
nohup ~/biosoft/sratoolkit/sratoolkit.2.6.3-centos_linux64/bin/fastq-dump --split-3 --gzip -A N080611_untreated SRR1039516.sra &
nohup ~/biosoft/sratoolkit/sratoolkit.2.6.3-centos_linux64/bin/fastq-dump --split-3 --gzip -A N080611_Dex SRR1039517.sra &
nohup ~/biosoft/sratoolkit/sratoolkit.2.6.3-centos_linux64/bin/fastq-dump --split-3 --gzip -A N080611_Alb SRR1039518.sra &
nohup ~/biosoft/sratoolkit/sratoolkit.2.6.3-centos_linux64/bin/fastq-dump --split-3 --gzip -A N080611_Alb_Dex SRR1039519.sra &
nohup ~/biosoft/sratoolkit/sratoolkit.2.6.3-centos_linux64/bin/fastq-dump --split-3 --gzip -A N061011_untreated SRR1039520.sra &
nohup ~/biosoft/sratoolkit/sratoolkit.2.6.3-centos_linux64/bin/fastq-dump --split-3 --gzip -A N061011_Dex SRR1039521.sra &
nohup ~/biosoft/sratoolkit/sratoolkit.2.6.3-centos_linux64/bin/fastq-dump --split-3 --gzip -A N061011_Alb SRR1039522.sra &
nohup ~/biosoft/sratoolkit/sratoolkit.2.6.3-centos_linux64/bin/fastq-dump --split-3 --gzip -A N061011_Alb_Dex SRR1039523.sra &
可以看到这里的16个样本来源于同样的4个人,是HASM细胞系,处理详情如下:
1) no treatment;2) treatment with a β2-agonist (i.e. Albuterol, 1μM for 18h);3) treatment with a glucocorticosteroid (i.e. Dexamethasone (Dex), 1μM for 18h);4) simultaneous treatment with a β2-agonist and glucocorticoid
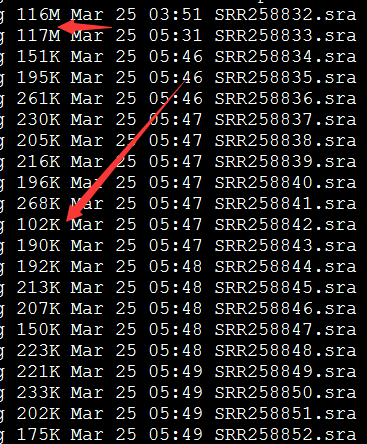
下载的sra大小如下:
-rw-rw-r-- 1 jmzeng jmzeng 1.6G Aug 9 04:21 SRR1039508.sra
-rw-rw-r-- 1 jmzeng jmzeng 1.5G Aug 9 05:20 SRR1039509.sra
-rw-rw-r-- 1 jmzeng jmzeng 1.6G Aug 9 06:14 SRR1039510.sra
-rw-rw-r-- 1 jmzeng jmzeng 1.5G Aug 9 07:05 SRR1039511.sra
-rw-rw-r-- 1 jmzeng jmzeng 2.1G Aug 9 08:07 SRR1039512.sra
-rw-rw-r-- 1 jmzeng jmzeng 2.3G Aug 9 09:17 SRR1039513.sra
-rw-rw-r-- 1 jmzeng jmzeng 3.1G Aug 9 10:56 SRR1039514.sra
-rw-rw-r-- 1 jmzeng jmzeng 1.9G Aug 9 11:56 SRR1039515.sra
-rw-rw-r-- 1 jmzeng jmzeng 2.1G Aug 9 13:02 SRR1039516.sra
-rw-rw-r-- 1 jmzeng jmzeng 2.6G Aug 9 14:16 SRR1039517.sra
-rw-rw-r-- 1 jmzeng jmzeng 2.3G Aug 9 15:17 SRR1039518.sra
-rw-rw-r-- 1 jmzeng jmzeng 2.0G Aug 9 16:05 SRR1039519.sra
-rw-rw-r-- 1 jmzeng jmzeng 2.1G Aug 9 16:56 SRR1039520.sra
-rw-rw-r-- 1 jmzeng jmzeng 2.4G Aug 9 17:57 SRR1039521.sra
-rw-rw-r-- 1 jmzeng jmzeng 2.0G Aug 9 18:46 SRR1039522.sra
-rw-rw-r-- 1 jmzeng jmzeng 1.4G Aug 9 19:28 SRR1039523.sra
解压后成双端测序的fastq数据如下:
-rw-rw-r-- 1 jmzeng jmzeng 2.5G Aug 9 20:12 N052611_Alb_1.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 2.5G Aug 9 20:12 N052611_Alb_2.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 1.3G Aug 9 20:44 N052611_Alb_Dex_1.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 1.3G Aug 9 20:44 N052611_Alb_Dex_2.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 289M Aug 9 20:44 N052611_Alb_Dex.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 951M Aug 9 20:59 N052611_Dex_1.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 954M Aug 9 20:59 N052611_Dex_2.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 1.7G Aug 9 20:53 N052611_untreated_1.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 1.7G Aug 9 20:53 N052611_untreated_2.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 1.5G Aug 9 20:45 N061011_Alb_1.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 1.5G Aug 9 20:45 N061011_Alb_2.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 1.9G Aug 9 20:59 N061011_Alb_Dex_1.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 1.9G Aug 9 20:59 N061011_Alb_Dex_2.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 16M Aug 9 20:45 N061011_Alb.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 1.4G Aug 9 20:48 N061011_Dex_1.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 1.4G Aug 9 20:48 N061011_Dex_2.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 1.2G Aug 9 20:00 N061011_untreated_1.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 1.2G Aug 9 20:00 N061011_untreated_2.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 759M Aug 9 20:00 N061011_untreated.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 1.9G Aug 9 20:03 N080611_Alb_1.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 1.9G Aug 9 20:03 N080611_Alb_2.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 1.3G Aug 9 19:59 N080611_Alb_Dex_1.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 1.3G Aug 9 19:59 N080611_Alb_Dex_2.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 535M Aug 9 19:59 N080611_Alb_Dex.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 2.1G Aug 9 20:06 N080611_Dex_1.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 2.1G Aug 9 20:06 N080611_Dex_2.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 1.6G Aug 9 20:01 N080611_untreated_1.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 1.6G Aug 9 20:01 N080611_untreated_2.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 1.3G Aug 9 08:09 N61311_Alb_1.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 1.3G Aug 9 08:09 N61311_Alb_2.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 1.3G Aug 9 08:08 N61311_Alb_Dex_1.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 1.3G Aug 9 08:08 N61311_Alb_Dex_2.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 1.2G Aug 9 08:07 N61311_Dex_1.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 1.2G Aug 9 08:07 N61311_Dex_2.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 1.3G Aug 9 08:09 N61311_untreated_1.fastq.gz
-rw-rw-r-- 1 jmzeng jmzeng 1.3G Aug 9 08:09 N61311_untreated_2.fastq.gz
接下来所有的分析就基于此数据啦