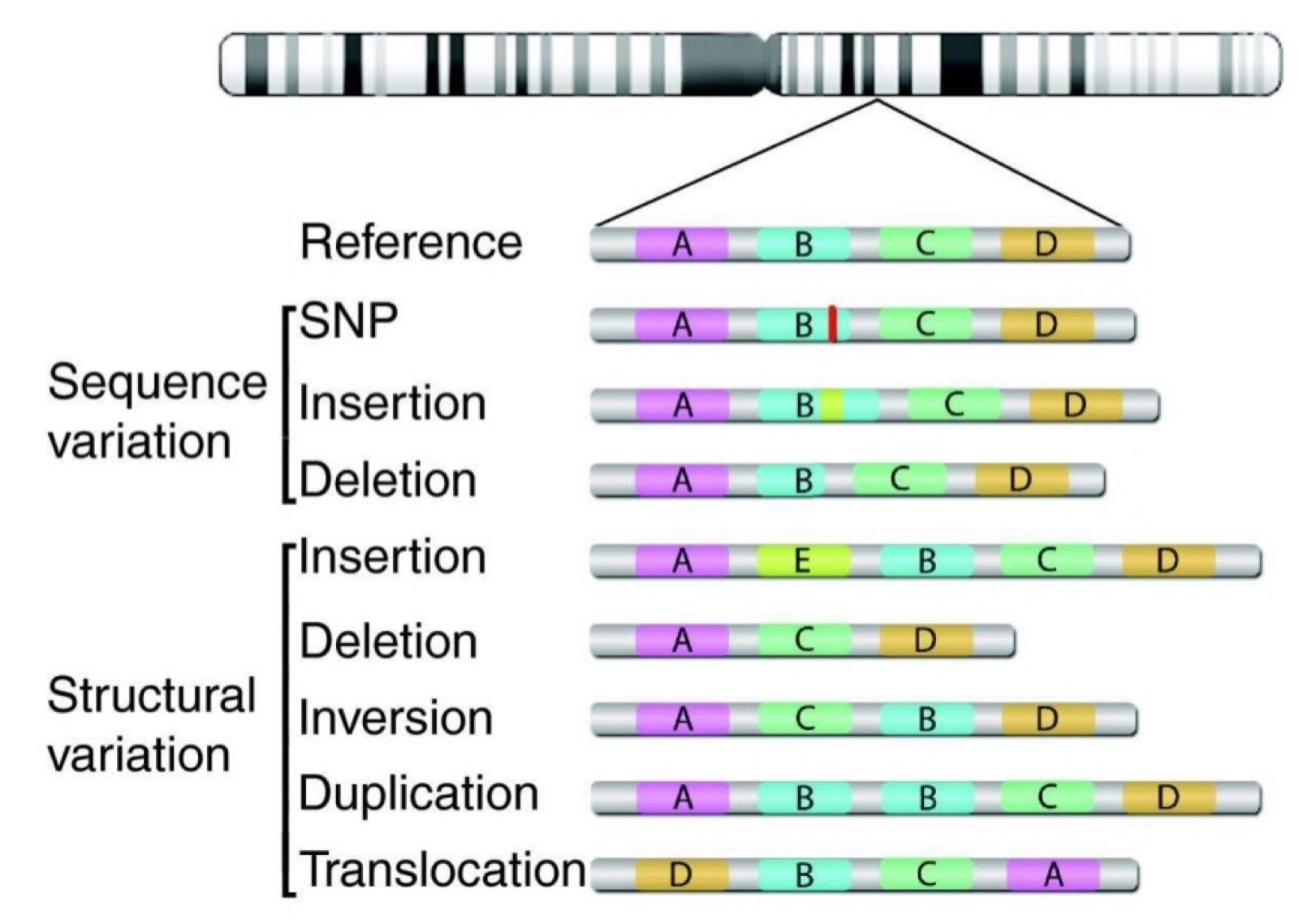
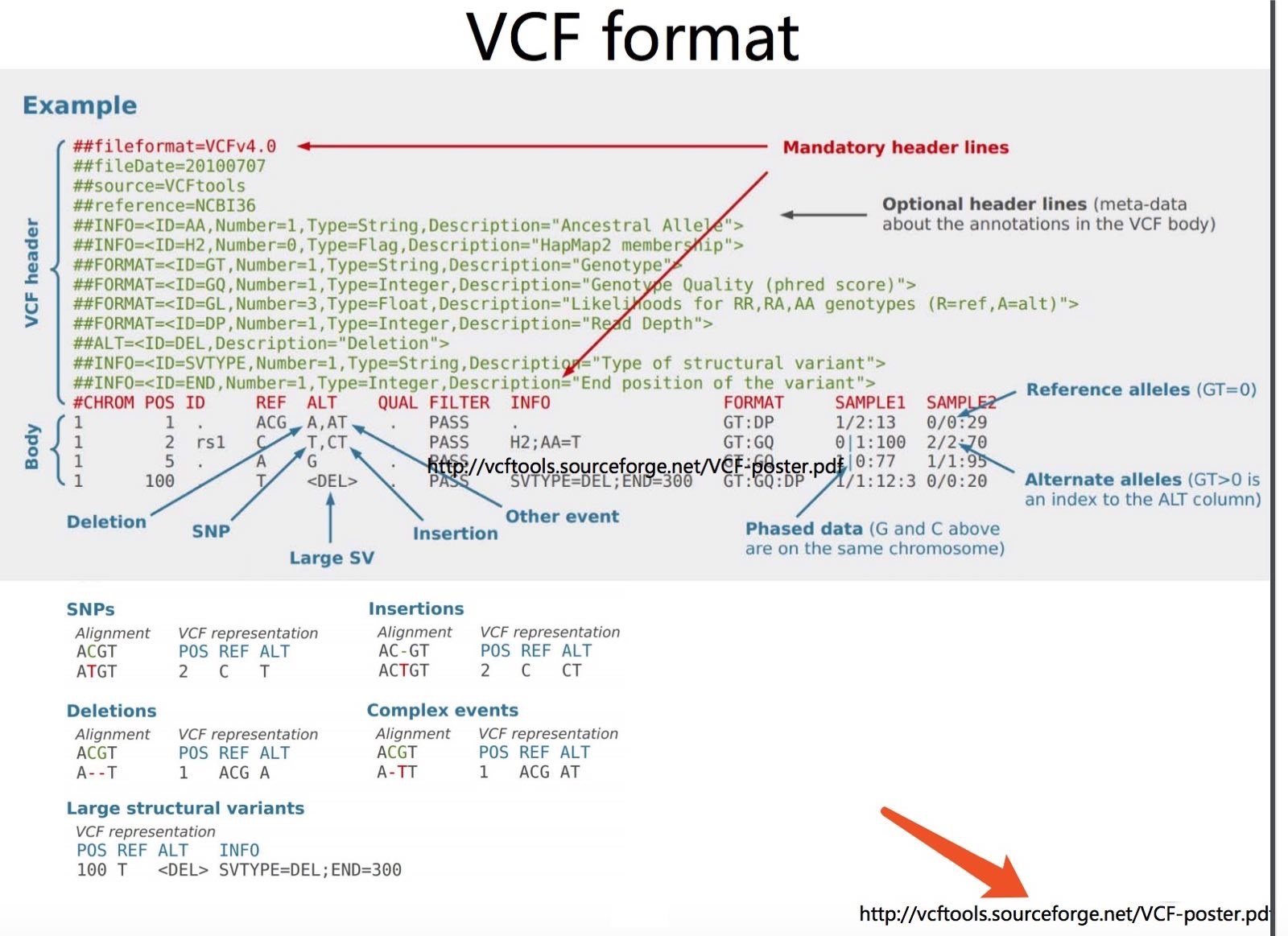
各行各业都有在自己的标准体系,生物信息学数据分析也不例外,各个厂商出品的芯片系列,还有各种NGS组学分析,都会涉及到不同的分析步骤,有着丰富多样的中间文件。其中一些常用的文件就被规定成文件格式。 文件格式那么多,都可以了解一二,当然,不需要背诵它们所有的细节,不过对下面我们单独拿出来详细介绍的还是尽量要耳熟能详。
简单来说,测序得到的是带有质量值的碱基序列(fastq格式),参加基因组是(fasta格式),用比对工具把fastq格式的序列回帖到对应的fasta格式的参考基因组序列,就可以产生sam格式的比对文件。 把sam格式的文本文件压缩成二进制的bam文件可以节省空间,如果对参考基因组上面的各个区段标记它们的性质,比如哪些区域是外显子,内含子,UTR等等,这就是gtf/gff格式。 如果只是为了单纯描述某个基因组区域,就是bed格式文件,记录染色体号以及起始终止坐标,正负链即可。 如果是记录某些位点或者区域碱基的变化,就是VCF文件格式。
FASTQ
FASTQ是基于文本的,保存生物序列(通常是核酸序列)和其测序质量信息的标准格式。 其序列以及质量信息都是使用一个ASCII字符标示,最初由Sanger开发。 目的是将FASTA序列与质量数据放到一起,目前已经成为高通量测序结果的实施标准。
定义和示例
FASTQ文件中每个序列通常有四行:
第一行是序列标识以及相关的描述信息,以‘@’开头
第二行是序列
第三行以‘+’开头,后面是序列标示符、描述信息,或者什么也不加,但是“+”不能少。
第四行,是质量信息,和第二行的序列相对应,每一个序列都有一个质量评分,根据评分体系的不同,每个字符的含义表示的数字也不相同。
一个简单的示例如下:
@SEQ_ID
GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT
+
!''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65
格式口诀:
*‘@’开头引标识*
*序列老二对老四*
*老三没“+”不好使*
*老四质量分数最多事*
序列标识
上面说到第一行是序列标识以及相关的描述信息,以‘@’开头。可以像上面的示例那么简单,但如果是正规测序仪下机的真实数据,通常会很复杂。比如:
@EAS139:136:FC706VJ:2:2104:15343:197393 1:Y:18:ATCACG
这个序列标识以及相关描述信息以冒号分割,每一个字段信息如下:
| EAS139 |
the unique instrument name |
| 136 |
the run id |
| FC706VJ |
the flowcell id |
| 2 |
flowcell lane |
| 2104 |
tile number within the flowcell lane |
| 15343 |
‘x’-coordinate of the cluster within the tile |
| 197393 |
‘y’-coordinate of the cluster within the tile |
| 1 |
the member of a pair, 1 or 2 (paired-end or mate-pair reads only) |
| Y |
Y if the read fails filter (read is bad), N otherwise |
| 18 |
0 when none of the control bits are on, otherwise it is an even number |
| ATCACG |
index sequence |
当然,上面的表格介绍的只是其中一个测序仪下机数据,如果是其它机器,产商可以自由定义标识符格式,因为fastq格式的第一行只需要以@符号开头即可。
不过,也有一些时候fastq数据并不是测序仪直接下机的,而且他人上传到了NCBI的SRA中心,我们下载下来解压后一般就没有了测序仪相关的标识,例子如下:
@SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36
GGGTGATGGCCGCTGCCGATGGCGTCAAATCCCACC
+SRR001666.1 071112_SLXA-EAS1_s_7:5:1:817:345 length=36
IIIIIIIIIIIIIIIIIIIIIIIIIIIIII9IG9IC
质量编码格式
质量评分指的是一个碱基的错误概率的对数值。 其最初在Phred拼接软件中定义与使用,其后在许多软件中得到使用。 其质量得分与错误概率的对应关系见下表:
| 10 |
1 in 10 |
90 % |
| 20 |
1 in 100 |
99% |
| 30 |
1 in 1000 |
99.9% |
| 40 |
1 in 10000 |
99.99% |
| 50 |
1 in 100000 |
99.999% |
Phred quality scores Q are defined as a property which is logarithmically related to the base-calling error probabilities P.
Q=-10lgP
除了Phred质量得分换算标准,还有就是Solexa标准:是把P换成p/(1-p)
对于每个碱基的质量编码标示,不同的软件采用不同的方案,目前有5种方案:
Sanger,Phred quality score,值的范围从0到92,对应的ASCII码从33到126,但是对于测序数据(raw read data)质量得分通常小于60,序列拼接或者mapping可能用到更大的分数。
Solexa/Illumina 1.0, Solexa/Illumina quality score,值的范围从-5到63,对应的ASCII码从59到126,对于测序数据,得分一般在-5到40之间。
Illumina 1.3+,Phred quality score,值的范围从0到62对应的ASCII码从64到126,低于测序数据,得分在0到40之间;
Illumina 1.5+,Phred quality score,但是0到2作为另外的标示。
Illumina 1.8+
下面是更为直观的表示:
SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS.....................................................
..........................XXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXXX......................
...............................IIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII......................
.................................JJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJJ......................
LLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLLL....................................................
!"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHIJKLMNOPQRSTUVWXYZ[\]^_`abcdefghijklmnopqrstuvwxyz{|}~
| | | | | |
33 59 64 73 104 126
S - Sanger Phred+33, raw reads typically (0, 40)
X - Solexa Solexa+64, raw reads typically (-5, 40)
I - Illumina 1.3+ Phred+64, raw reads typically (0, 40)
J - Illumina 1.5+ Phred+64, raw reads typically (3, 40)
with 0=unused, 1=unused, 2=Read Segment Quality Control Indicator (bold)
(Note: See discussion above).
L - Illumina 1.8+ Phred+33, raw reads typically (0, 41)
来至于 wikipedia:
文件后缀
没有特别的规定,通常使用.fq, .fastq, .txt等。 但是要注意,这个文件格式主要指的是文本文件里面的每行每列的内容规则,并不是我们常见的计算机领域的mp3,mp4,avi,xls,doc等等。
其它注意事项:
- 双端测序一般有两个文件(也可通过某种规则把两个文件合并成一个)。
- 第一个文件与第二个文件的行数完全一样,且测序序列的排列顺序完全一致。
- 在第一个文件中,描述信息的结尾是“/1”,表示是双端测序的一端;第二个文件中同样位置/行数的相对应的测序序列的描述信息则以“/2”结尾,表示是双端测序的另一端。(2.2.2的表2-5中有叙述)
参考链接:
https://en.wikipedia.org/wiki/FASTQ_format
SAM格式
SAM是一种序列比对格式标准,由sanger制定,是以TAB为分割符的文本格式。 主要应用于测序序列mapping到基因组上的结果表示,当然也可以表示任意的多重比对结果。 SAM的全称是sequence alignment/map format。
定义和示例
SAM分为两部分,注释信息(header section )和比对结果部分 (alignment section)。 通常是把FASTQ文件格式的测序数据比对到对应的参考基因组版本得到的。 注释信息并不是SAM文件的重点,是该SAM文件产生以及被处理过程的一个记录,规定以@开头,用不同的tag表示不同的信息,主要有:
- @HD,说明符合标准的版本、对比序列的排列顺序;
- @SQ,参考序列说明;
- @RG,比对上的序列(read)说明;
- @PG,使用的程序说明;
- @CO,任意的说明信息。
一个简单的SAM文件例子如下:
@HD VN:1.0 SO:unsorted
@SQ SN:chr1 LN:249250621
@SQ SN:chr2 LN:243199373
@PG ID:Bowtie VN:1.0.0 CL:"bowtie genome/hg19 -q reads/SRR3101251.fastq -m 1 -p 4 -S"
SRR3101251.1 0 chr19 9486878 255 49M * 0 0 NTACTCCCACTACTCTCAGATTCAAGCAATCCTCCCACCCTAGCCCACC #1=DDDFFHHHHHIHHIJJJHIJIIJIHIFHJIIJJJJJJJIIJJJJJJ XA:i:1 MD:Z:0A48 NM:i:1
SRR3101251.5 16 chr2 240279787 255 49M * 0 0 CCTGAATCCATCAGAGCAGCCGGGCTGTGACACTCACTGTCATGATGTT JIJJIHIIIIJJJJJJJJJGHJJJJIIHJHICJIGCHHHHHFFFFFCCC XA:i:0 MD:Z:49 NM:i:0
SRR3101251.6 4 * 0 0 * * 0 0 NATTCCCACCTATGAGTGAGAATATGCGGTGTTTGGTTTTTTGTTCTTG #1=DDDFFHHHHHJJJGHIJJJJJJJJJJCGGIIJJIIJJJIJHJIIJJ XM:i:1
前四行是注释信息,后面是比对结果,下面对比对结果进行解释,它是SAM格式文件的精华部分。
比对结果详解
SRR035022.2621862 163 16 59999 37 22S54M = 60102 179 CCAACCCAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCGACCCTCACCCTCACCC >AAA=>?AA>@@B@B?AABAB?AABAB?AAC@B?@AB@A?A>A@A?AAAAB??ABAB?79A?AAB;B?@?@<=8:8 XT:A:M XN:i:2 SM:i:37 AM:i:37 XM:i:0 XO:i:0 XG:i:0 RG:Z:SRR035022 NM:i:2 MD:Z:0N0N52 OQ:Z:CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCBCCCCCCBBCC@CCCCCCCCCCACCCCC;CCCBBC?CCCACCACA@
所以在我们的例子中,每一个字段的说明如下:
QNAME SRR035022.2621862
FLAG 163
RNAME 16
POS 59999
MAQ 37
CIGAR 22S54M
MRNM =
MPOS 60102
ISIZE 179
SEQ CCAACCCAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCGACCCTCACCCTCACCC
QUAL >AAA=>?AA>@@B@B?AABAB?AABAB?AAC@B?@AB@A?A>A@A?AAAAB??ABAB?79A?AAB;B?@?@<=8:8
TAG XT:A:M
TAG XN:i:2
TAG SM:i:37
TAG AM:i:37
TAG XM:i:0
TAG XO:i:0
TAG XG:i:0
TAG RG:Z:SRR035022
TAG NM:i:2
TAG MD:Z:0N0N52
TAG OQ:Z:CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCBCCCCCCBBCC@CCCCCCCCCCACCCCC;CCCBBC?CCCACCACA
比对结果部分(alignment section),每一行表示一个片段(segment)的比对信息,包括11个必须的字段(mandatory fields)和一个可选的字段,字段之间用tag分割。
必须的字段有11个,顺序固定,不可用时,根据字段定义,可以为’0‘或者’*’,这是11个字段包括:
QNAME,比对片段的(template)的编号;
FLAG,位标识,template mapping情况的数字表示,每一个数字代表一种比对情况,这里的值是符合情况的数字相加总和; (picard专门有一个工具解读sam的flag:http://broadinstitute.github.io/picard/explain-flags.html)
1 The read is one of a pair read是pair中的一条(read表示本条read,mate表示pair中的另一条read)
2 The alignment is one end of a proper paired-end alignment pair一正一负完美的比对上
4 The read has no reported alignments 这条read没有比对上
8 The read is one of a pair and has no reported alignments mate没有比对上
16 The alignment is to the reverse reference strand 这条read反向比对
32 The other mate in the paired-end alignment is aligned to the reverse reference strand mate反向比对
64 The read is the first (#1) mate in a pair 这条read是read1
128 The read is the second (#2) mate in a pair 这条read是read2
RNAME,参考序列的编号,如果注释中对SQ-SN进行了定义,这里必须和其保持一致,另外对于没有mapping上的序列,这里是’*’;
POS,比对上的位置,注意是从1开始计数,没有比对上,此处为0;
MAPQ,mappint的质量;
CIGAR,简要比对信息表达式(Compact Idiosyncratic Gapped Alignment Report),其以参考序列为基础,使用数字加字母表示比对结果,比如3S6M1P1I4M,前三个碱基被剪切去除了,然后6个比对上了,然后打开了一个缺口,有一个碱基插入,最后是4个比对上了,是按照顺序的;
M”表示 match或 mismatch;
“I”表示 insert;
“D”表示 deletion;
“N”表示 skipped(跳过这段区域);
“S”表示 soft clipping(被剪切的序列存在于序列中);
“H”表示 hard clipping(被剪切的序列不存在于序列中);
“P”表示 padding;
“=”表示 match;
“X”表示 mismatch(错配,位置是一一对应的);
RNEXT,下一个片段比对上的参考序列的编号,没有另外的片段,这里是’*‘,同一个片段,用’=’;
PNEXT,下一个片段比对上的位置,如果不可用,此处为0;
TLEN,Template的长度,最左边得为正,最右边的为负,中间的不用定义正负,不分区段(single-segment)的比对上,或者不可用时,此处为0;
SEQ,序列片段的序列信息,如果不存储此类信息,此处为’*’,注意CIGAR中M/I/S/=/X对应数字的和要等于序列长度;
QUAL,序列的质量信息,格式同FASTQ一样。read质量的ASCII编码。
可选字段(optional fields),格式如:TAG:TYPE:VALUE,其中TAG有两个大写字母组成,每个TAG代表一类信息,每一行一个TAG只能出现一次,TYPE表示TAG对应值的类型,可以是字符串、整数、字节、数组等。
SAM要处理好的问题
- 非常多序列(read),mapping到多个参考基因组(reference)上
- 同一条序列,分多段(segment)比对到参考基因组上
- 无限量的,结构化信息表示,包括错配、删除、插入等比对信息
要注意的几个概念,以及与之对应的模型:
- reference
- read
- segment
- template(参考序列和比对上的序列共同组成的序列为template)
- alignment
- seq
参考链接:
https://en.wikipedia.org/wiki/SAM_(file_format)
VCF
Variant Call Format(VCF)是一个用于存储基因序列突变信息的文本格式。 可以表示单碱基突变, 插入/缺失, 拷贝数变异和结构变异等。 通常是对BAM文件格式的比对结果进行处理得到的。 BCF格式文件是VCF格式的二进制文件。我们就不再介绍BCF格式啦。
提到vcf就必须提到千人基因组计划,因为千人计划组才产生的vcf。生信菜鸟团有一篇博客《居然可以下载千人基因组计划的所有数据bam,vcf数据》 http://www.bio-info-trainee.com/1339.html 专门讲了千人计划的数据下载。
当然,所有的数据格式定义,都推荐大家看原汁原味的英文介绍,那个是金标准,我们的翻译只是为了促进大家的理解,如果有模棱两可的地方,以英文原文为准:https://samtools.github.io/hts-specs/VCFv4.2.pdf
定义和示例
如上图所示,vcf记录的即为各类型的变异。例如:点突变,拷贝数变异,插入,缺失等结构变异。
VCF分为两部分,注释信息和变异位点记录信息。
注释信息通常以#开头,会描述该VCF版本,目前以4.2居多,然后会一行行记录变异位点信息里面会出现的所有TAG。
下面这个是NCBI的dbSNP数据库里面的人类的vcf文件的部分截取:
##fileformat=VCFv4.0
##fileDate=20160601
##source=dbSNP
##dbSNP_BUILD_ID=147
##reference=GRCh37.p13
##phasing=partial
##variationPropertyDocumentationUrl=ftp://ftp.ncbi.nlm.nih.gov/snp/specs/dbSNP_BitField_latest.pdf
##INFO=<ID=RS,Number=1,Type=Integer,Description="dbSNP ID (i.e. rs number)">
##INFO=<ID=RSPOS,Number=1,Type=Integer,Description="Chr position reported in dbSNP">
##INFO=<ID=RV,Number=0,Type=Flag,Description="RS orientation is reversed">
#CHROM POS ID REF ALT QUAL FILTER INFO
1 10177 rs201752861 A C . . RS=201752861;RSPOS=10177;dbSNPBuildID=137;SSR=0;SAO=0;VP=0x050000020005000002000100;GENEINFO=DDX11L1:100287102;WGT=1;VC=SNV;R5;ASP
1 10177 rs367896724 A AC . . RS=367896724;RSPOS=10177;dbSNPBuildID=138;SSR=0;SAO=0;VP=0x050000020005170026000200;GENEINFO=DDX11L1:100287102;WGT=1;VC=DIV;R5;ASP;VLD;G5A;G5;KGPhase3;CAF=0.5747,0.4253;COMMON=1
1 10228 rs143255646 TA T . . RS=143255646;RSPOS=10229;dbSNPBuildID=134;SSR=0;SAO=0;VP=0x050000020005000002000200;GENEINFO=DDX11L1:100287102;WGT=1;VC=DIV;R5;ASP
1 10228 rs200462216 TAACCCCTAACCCTAACCCTAAACCCTA T . . RS=200462216;RSPOS=10229;dbSNPBuildID=137;SSR=0;SAO=0;VP=0x050000020005000002000200;GENEINFO=DDX11L1:100287102;WGT=1;VC=DIV;R5;ASP
1 10230 rs775928745 AC A . . RS=775928745;RSPOS=10231;dbSNPBuildID=144;SSR=0;SAO=0;VP=0x050000020005000002000200;GENEINFO=DDX11L1:100287102;WGT=1;VC=DIV;R5;ASP
1 10231 rs200279319 C A . . RS=200279319;RSPOS=10231;dbSNPBuildID=137;SSR=0;SAO=0;VP=0x050000020005000002000100;GENEINFO=DDX11L1:100287102;WGT=1;VC=SNV;R5;ASP
1 10234 rs145599635 C T . . RS=145599635;RSPOS=10234;dbSNPBuildID=134;SSR=0;SAO=0;VP=0x050100020005000002000100;GENEINFO=DDX11L1:100287102;WGT=1;VC=SNV;SLO;R5;ASP
1 10235 rs540431307 T TA . . RS=540431307;RSPOS=10235;dbSNPBuildID=142;SSR=0;SAO=0;VP=0x050000020005040024000200;GENEINFO=DDX11L1:100287102;WGT=1;VC=DIV;R5;ASP;VLD;KGPhase3;CAF=0.9988,0.001198;COMMON=0
1 10247 rs796996180 T C . . RS=796996180;RSPOS=10247;dbSNPBuildID=146;SSR=0;SAO=0;VP=0x050100020005000002000100;GENEINFO=DDX11L1:100287102;WGT=1;VC=SNV;SLO;R5;ASP
限于文章篇幅限制,我只是截取了该VCF文件的部分注释信息,很明显可以看到注释信息刚刚开始的几行其实是没有规则的,只需要以##开头即可,描述一些必备信息,包括参考基因组版本,得到该VCF文件的命令是什么等等。
后面的都是以INFO=<ID=······>的形式来介绍一个个TAG,这些TAG都是会在VCF的正文,变异位点记录里面用到的。而且每个tag都很容易理解,就是对应的英文描述而已。
接下来我们看看比较复杂的正文部分,就是变异位点记录信息。
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT sample1
1 858691 . TG T 222 . INDEL;IDV=37;IMF=0.486842;DP=76;VDB=0.110516;SGB=-0.693139;MQSB=1;MQ0F=0;ICB=1;HOB=0.5;AC=1;AN=2;DP4=12,24,14,22;MQ=60 GT:PL 0/1:255,0,255
1 858801 . A G 222 . DP=59;VDB=0.728126;SGB=-0.692717;RPB=0.748623;MQB=1;MQSB=1;BQB=0.963908;MQ0F=0;ICB=1;HOB=0.5;AC=1;AN=2;DP4=14,17,11,12;MQ=60 GT:PL 0/1:255,0,255
1 859404 . C G 222 . DP=81;VDB=0.0896228;SGB=-0.693132;RPB=0.849598;MQB=1;MQSB=1;BQB=0.486963;MQ0F=0;ICB=1;HOB=0.5;AC=1;AN=2;DP4=17,15,18,16;MQ=60 GT:PL 0/1:255,0,255
1 859690 . C G 222 . DP=75;VDB=0.0662538;SGB=-0.69312;RPB=0.959181;MQB=1;MQSB=1;BQB=0.962588;MQ0F=0;ICB=1;HOB=0.5;AC=1;AN=2;DP4=20,15,18,14;MQ=60 GT:PL 0/1:255,0,255
1 859701 . C G 222 . DP=74;VDB=0.274853;SGB=-0.693127;RPB=0.97201;MQB=1;MQSB=1;BQB=0.717302;MQ0F=0;ICB=1;HOB=0.5;AC=1;AN=2;DP4=19,15,19,14;MQ=60 GT:PL 0/1:255,0,255
1 859913 . A G 222 . DP=67;VDB=0.756546;SGB=-0.693139;RPB=0.950685;MQB=1;MQSB=1;BQB=0.662934;MQ0F=0;ICB=1;HOB=0.5;AC=1;AN=2;DP4=18,10,19,17;MQ=60 GT:PL 0/1:255,0,255
1 860416 . G A 222 . DP=79;VDB=0.673886;SGB=-0.693144;RPB=0.11919;MQB=1;MQSB=1;BQB=0.992984;MQ0F=0;ICB=1;HOB=0.5;AC=1;AN=2;DP4=18,15,24,15;MQ=60 GT:PL 0/1:255,0,255
每一行代表一个Variant的信息。
CHROM 和 POS:代表参考序列名和variant的位置;如果是INDEL的话,位置是INDEL的第一个碱基位置。
ID:variant的ID。比如在dbSNP中有该SNP的id,则会在此行给出(这个需要自己下载dbSNP数据库文件进行注释才有的)。 若没有或者注释不上,则用’.’表示其为一个novel variant。
REF 和 ALT:参考序列的碱基 和 Variant的碱基。
QUAL:Phred格式(Phred_scaled)的质量值,表示在该位点存在variant的可能性;该值越高,则variant的可能性越大; 计算方法:Phred值 = -10 * log (1-p) p为variant存在的概率; 通过计算公式可以看出值为10的表示错误概率为0.1,该位点为variant的概率为90%。
FILTER:使用上一个QUAL值来进行过滤的话,是不够的。GATK能使用其它的方法来进行过滤,过滤结果中通过则该值为”PASS”;若variant不可靠,则该项不为”PASS”或”.”。
INFO: 这一行是variant的详细信息,内容很多,以下再具体详述。
FORMAT 和 sample1 :这两行合起来提供了 sample1 这个sample的基因型的信息。’sample1′代表这该名称的样品,是由SAM/BAM文件中的@RG下的 SM 标签决定的。 (当然并不是所有的VCF都是由一个BAM文件产生,比如数据库dbSNP提供的vcf文件,就没有样本信息啦)
第8列的INFO
该列信息最多了,都是以 “TAG=Value”, 并使用”;”分隔的形式 。 其中 很多的注释信息在VCF文件的头部注释中给出。以下是这些TAG的解释
AC,AF 和 AN:AC(Allele Count) 表示该Allele的数目;AF(Allele Frequency) 表示Allele的频率; AN(Allele Number) 表示Allele的总数目。对于1个diploid sample而言:则基因型 0/1 表示sample为杂合子,Allele数为1(双倍体的sample在该位点只有1个等位基因发生了突变),Allele的频率为0.5(双倍体的 sample在该位点只有50%的等位基因发生了突变),总的Allele为2; 基因型 1/1 则表示sample为纯合的,Allele数为2,Allele的频率为1,总的Allele为2。
DP:reads覆盖度。是一些reads被过滤掉后的覆盖度。
Dels:Fraction of Reads Containing Spanning Deletions。进行SNP和INDEL calling的结果中,有该TAG并且值为0表示该位点为SNP,没有则为INDEL。
FS:使用Fisher’s精确检验来检测strand bias而得到的Fhred格式的p值。该值越小越好。一般进行filter的时候,可以设置 FS < 10~20。
HaplotypeScore:Consistency of the site with at most two segregating haplotypes
InbreedingCoeff:Inbreeding coefficient as estimated from the genotype likelihoods per-sample when compared against the Hard-Weinberg expectation
MLEAC:Maximum likelihood expectation (MLE) for the allele counts (not necessarily the same as the AC), for each ALT allele, in the same order as listed
MLEAF:Maximum likelihood expectation (MLE) for the allele frequency (not necessarily the same as the AF), for each ALT alle in the same order as listed
MQ:RMS Mapping Quality
MQ0:Total Mapping Quality Zero Reads
MQRankSum:Z-score From Wilcoxon rank sum test of Alt vs. Ref read mapping qualities
QD:Variant Confidence/Quality by Depth
RPA:Number of times tandem repeat unit is repeated, for each allele (including reference)
RU:Tandem repeat unit (bases)
ReadPosRankSum:Z-score from Wilcoxon rank sum test of Alt vs. Ref read position bias
STR:Variant is a short tandem repeat
9和10列代表基因型
GT:AD:DP:GQ:PL 0/1:173,141:282:99:255,0,255
GT:AD:DP:GQ:PL 0/1:1,3:4:25.92:103,0,26
看上面最后两列数据,这两列数据是对应的,前者为格式,后者为格式对应的数据。这些TAG也是可以在VCF的头文件找到的
GT:样品的基因型(genotype)。两个数字中间用’/’分开,这两个数字表示双倍体的sample的基因型。0 表示样品中有ref的allele; 1 表示样品中variant的allele; 2表示有第二个variant的allele。因此: 0/0 表示sample中该位点为纯合的,和ref一致; 0/1 表示sample中该位点为杂合的,有ref和variant两个基因型; 1/1 表示sample中该位点为纯合的,和variant一致。
AD 和 DP:AD(Allele Depth)为sample中每一种allele的reads覆盖度,在diploid中则是用逗号分割的两个值,前者对应ref基因型,后者对应variant基因型;
DP(Depth)为sample中该位点的测序深度。
GQ:基因型的质量值(Genotype Quality)。Phred格式(Phred_scaled)的质量值,表示在该位点该基因型存在的可能性;该值越高,则Genotype的可能性越大;计算方法:Phred值 = -10 * log (1-p) p为基因型存在的概率。
PL:指定的三种基因型的质量值(provieds the likelihoods of the given genotypes)。这三种指定的基因型为(0/0,0/1,1/1),这三种基因型的概率总和为1。和之前不一致,该值越大,表明为该种基因型的可能性越小。 Phred值 = -10 * log (p) p为基因型存在的概率。
上图可以帮助很好的理解vcf格式。
注意事项:
针对vcf格式有如bcftools等软件进行处理。
生信菜鸟团的博客和生信技能树里面都介绍了很多处理vcf的软件。
强烈推荐去看《生信技能树》中的:我的基因组28 https://en.wikipedia.org/wiki/Variant_Call_Format
GTF和GFF
GFF全称为general feature format,这种格式主要是用来注释基因组。
GTF全称为gene transfer format,主要是用来对基因进行注释。
GTF和GFF格式是Sanger研究所定义,是一种简单的、方便的对于DNA、RNA以及蛋白质序列的特征进行描述的一种数据格式. 比如序列的哪里到哪里是基因,是转录本,是外显子,内含子或者CDS等等,已经成为序列注释的通用格式,许多软件都支持输入或者输出gff格式。
定义和示例
gff由tab键隔开的9列组成,以下是各列的说明:
Column 1: “seqid”
序列的编号,编号的有效字符[a-zA-Z0-9.:^*$@!+_?-|]
Column 2: “source”
注释信息的来源,比如”Genescan”、”Genbank” 等,可以为空,为空用”.”点号代替
Column 3: “type”
注释信息的类型,比如Gene、cDNA、mRNA等,或者是SO对应的编号
Columns 4 & 5: “start” and “end”
开始与结束的位置,注意计数是从1开始的。结束位置不能大于序列的长度
Column 6: “score”
得分,数字,是注释信息可能性的说明,可以是序列相似性比对时的E-values值或者基因预测是的P-values值。”.”表示为空。
Column 7: “strand”
序列的方向, +表示正义链, -反义链 , ? 表示未知.
Column 8: “phase”
仅对注释类型为 “CDS”有效,表示起始编码的位置,有效值为0、1、2。
Column 9: “attributes”
以多个键值对组成的注释信息描述,键与值之间用”=“,不同的键值用”;“隔开,一个键可以有多个值,不同值用”,“分割。注意如果描述中包括tab键以及”,=;”,要用URL转义规则进行转义,如tab键用 %09代替。键是区分大小写的,以大写字母开头的键是预先定义好的,在后面可能被其他注释信息所调用。
预先定义的键包括:
ID 注释信息的编号,在一个GFF文件中必须唯一;
Name 注释信息的名称,可以重复;
Alias 别名
Parent Indicates 该注释所属的注释,值为注释信息的编号,比如外显子所属的转录组编号,转录组所属的基因的编号。值可以为多个。
Target Indicates: the target of a nucleotide-to-nucleotide or protein-to-nucleotide alignment.(核苷酸对核苷酸或蛋白质至核苷酸比对的靶点。)
Gap:The alignment of the feature to the target if the two are not collinear (e.g. contain gaps).(如果两者不共线(例如包含间隙),则该特征与目标的对准)
Derives_from:Used to disambiguate the relationship between one feature and another when the relationship is a temporal one rather than a purely structural “part of” one. This is needed for polycistronic genes.(用于消除一个特征与另一个特征之间的关系,当关系是一个时间的关系,而不是纯粹的结构“一部分”时。 这是多顺反子基因所必需的。)
Note: 备注
Dbxref :数据库索引
Ontology_term: A cross reference to an ontology term.
下面是一个简单的实例:
##gff-version 3
##sequence-region ctg123 1 1497228
ctg123 . gene 1000 9000 . + . ID=gene00001;Name=EDEN
ctg123 . TF_binding_site 1000 1012 . + . Parent=gene00001
ctg123 . mRNA 1050 9000 . + . ID=mRNA00001;Parent=gene00001
ctg123 . mRNA 1050 9000 . + . ID=mRNA00002;Parent=gene00001 ctg123 . mRNA 1300 9000 . + . ID=mRNA00003;Parent=gene00001 ctg123 . exon 1300 1500 . + . Parent=mRNA00003
ctg123 . exon 1050 1500 . + . Parent=mRNA00001,mRNA00002
ctg123 . exon 3000 3902 . + . Parent=mRNA00001,mRNA00003
ctg123 . exon 5000 5500 . + . Parent=mRNA00001,mRNA00002,mRNA00003
ctg123 . exon 7000 9000 . + . Parent=mRNA00001,mRNA00002,mRNA00003
ctg123 . CDS 1201 1500 . + 0 ID=cds00001;Parent=mRNA00001
ctg123 . CDS 3000 3902 . + 0 ID=cds00001;Parent=mRNA00001
ctg123 . CDS 5000 5500 . + 0 ID=cds00001;Parent=mRNA00001
ctg123 . CDS 7000 7600 . + 0 ID=cds00001;Parent=mRNA00001
ctg123 . CDS 1201 1500 . + 0 ID=cds00002;Parent=mRNA00002
ctg123 . CDS 5000 5500 . + 0 ID=cds00002;Parent=mRNA00002
ctg123 . CDS 7000 7600 . + 0 ID=cds00002;Parent=mRNA00002
ctg123 . CDS 3301 3902 . + 0 ID=cds00003;Parent=mRNA00003
ctg123 . CDS 5000 5500 . + 2 ID=cds00003;Parent=mRNA00003
ctg123 . CDS 7000 7600 . + 2 ID=cds00003;Parent=mRNA00003
ctg123 . CDS 3391 3902 . + 0 ID=cds00004;Parent=mRNA00003
ctg123 . CDS 5000 5500 . + 2 ID=cds00004;Parent=mRNA00003
ctg123 . CDS 7000 7600 . + 2 ID=cds00004;Parent=mRNA00003
可以看到第9列其实是可以无限扩展的,只需要以封号进行分割即可。
GTF 与GFF的差异
GTF文件以及GFF文件都由9列数据组成,这两种文件的前8列都是相同的(一些小的差别),它们的差别重点在第9列。
GTF文件的第9列同GFF文件不同,虽然同样是标签与值配对的情况,但标签与值之间以空格分开,而不是GFF里面的=号 且每个特征之后都要有分号,(包括最后一个特征). 下面看一个GTF的实例:
17 havana five_prime_utr 7687377 7687427 . - . gene_id "ENSG00000141510"; gene_version "16"; transcript_id "ENST00000503591"; transcript_version "1"; gene_name "TP53"; gene_source "ensembl_havana"; gene_biotype "protein_coding"; havana_gene "OTTHUMG00000162125"; havana_gene_version "10"; transcript_name "TP53-003"; transcript_source "havana"; transcript_biotype "protein_coding"; havana_transcript "OTTHUMT00000367399"; havana_transcript_version "2"; tag "cds_end_NF"; tag "mRNA_end_NF"; transcript_support_level "5";
17 havana five_prime_utr 7677325 7677427 . - . gene_id "ENSG00000141510"; gene_version "16"; transcript_id "ENST00000503591"; transcript_version "1"; gene_name "TP53"; gene_source "ensembl_havana"; gene_biotype "protein_coding"; havana_gene "OTTHUMG00000162125"; havana_gene_version "10"; transcript_name "TP53-003"; transcript_source "havana"; transcript_biotype "protein_coding"; havana_transcript "OTTHUMT00000367399"; havana_transcript_version "2"; tag "cds_end_NF"; tag "mRNA_end_NF"; transcript_support_level "5";
17 havana five_prime_utr 7676595 7676622 . - . gene_id "ENSG00000141510"; gene_version "16"; transcript_id "ENST00000503591"; transcript_version "1"; gene_name "TP53"; gene_source "ensembl_havana"; gene_biotype "protein_coding"; havana_gene "OTTHUMG00000162125"; havana_gene_version "10"; transcript_name "TP53-003"; transcript_source "havana"; transcript_biotype "protein_coding"; havana_transcript "OTTHUMT00000367399"; havana_transcript_version "2"; tag "cds_end_NF"; tag "mRNA_end_NF"; transcript_support_level "5";
速记口诀
* GTF,GFF 9列数据来组成*
*结果前8列都相同*
*GFF 9列间以tab 来分割,第9列用等号*
*GTF不服气,标签与值用空格分离,特征之间也用分号分开;要问特立独行谁最牛,GTF排第一!*
有趣小故事:
从前双胞胎兄弟,GTF,GFF。GFF是哥哥,GTF是弟弟,性别相同,都是由用来注释基因的,都是9个器官~~哥哥呢,比较随和,哥哥器官就用tab分开,然后第九个一对就划等号!
GTF是弟弟,天天心里想”WTF“! 不服气,就要特立独行!然后不服气,9个器官有八个都与哥哥相同,第9个子集偏不,就用了空格把它分成两块~~各个器官之间也是用分号分开!!预示子集跟各个不一样,自己的独立性!
目前两种文件可以方便的相互转化,比如:使用Cufflinks软件的 的gffread。
参考链接 http://genome.ucsc.edu/goldenPath/help/customTrack.html#GTF http://blog.sina.com.cn/s/blog_8a4f556e0102yd3l.html
GenePred
这种格式主要用在基因浏览器中基因预测的track。如果有可变剪切的情况,那表格的每一行就是一个 transcript 的全部信息。
定义和示例
每一行的具体解释如下
table genePred
"A gene prediction."
(
string name; "Name of gene"
string chrom; "Chromosome name"
char[1] strand; "+ or - for strand"
uint txStart; "Transcription start position"
uint txEnd; "Transcription end position"
uint cdsStart; "Coding region start"
uint cdsEnd; "Coding region end"
uint exonCount; "Number of exons"
uint[exonCount] exonStarts; "Exon start positions"
uint[exonCount] exonEnds; "Exon end positions"
)
如果觉得抽象,我们可以用示例来进行一下对比。小编在这里首先将模式植物拟南芥的gtf文件转化为gpd格式。head -n 1 看一下gpd文件第一行的样式。
#gpd
AT1G01010.1 Chr1 + 3630 5899 3759 5630 6 3630,3995,4485,4705,5173,5438, 3913,4276,4605,5095,5326,5899
再来grep一下gtf文件中有AT1G01010.1信息的那些行是什么样
#gtf
Chr1 Araport11 5UTR 3631 3759 . + . transcript_id "AT1G01010.1"; gene_id "AT1G01010"
Chr1 Araport11 exon 3631 3913 . + . transcript_id "AT1G01010.1"; gene_id "AT1G01010"
Chr1 Araport11 start_codon 3760 3762 . + . transcript_id "AT1G01010.1"; gene_id "AT1G01010"
Chr1 Araport11 CDS 3760 3913 . + 0 transcript_id "AT1G01010.1"; gene_id "AT1G01010"
Chr1 Araport11 exon 3996 4276 . + . transcript_id "AT1G01010.1"; gene_id "AT1G01010"
Chr1 Araport11 CDS 3996 4276 . + 2 transcript_id "AT1G01010.1"; gene_id "AT1G01010"
Chr1 Araport11 exon 4486 4605 . + . transcript_id "AT1G01010.1"; gene_id "AT1G01010"
Chr1 Araport11 CDS 4486 4605 . + 0 transcript_id "AT1G01010.1"; gene_id "AT1G01010"
Chr1 Araport11 exon 4706 5095 . + . transcript_id "AT1G01010.1"; gene_id "AT1G01010"
Chr1 Araport11 CDS 4706 5095 . + 0 transcript_id "AT1G01010.1"; gene_id "AT1G01010"
Chr1 Araport11 exon 5174 5326 . + . transcript_id "AT1G01010.1"; gene_id "AT1G01010"
Chr1 Araport11 CDS 5174 5326 . + 0 transcript_id "AT1G01010.1"; gene_id "AT1G01010"
Chr1 Araport11 CDS 5439 5630 . + 0 transcript_id "AT1G01010.1"; gene_id "AT1G01010"
Chr1 Araport11 exon 5439 5899 . + . transcript_id "AT1G01010.1"; gene_id "AT1G01010"
Chr1 Araport11 stop_codon 5628 5630 . + . transcript_id "AT1G01010.1"; gene_id "AT1G01010"
Chr1 Araport11 3UTR 5631 5899 . + . transcript_id "AT1G01010.1"; gene_id "AT1G01010"
我们可以看出,在GTF中,AT1G01010.1这个transcript共有6个CDS,那么对应到相应gpd文件AT1G01010.1这一行的第8列就是6,而第9列和第10列则是这6个CDS对应的起始和终止位置。
细心的朋友可能会发现,GTF文件中CDS起始位置在GenePred table中统统少了1,这其实就是两种文件的起始位置从1开始还是从0开始计数的区别。
GTF的产生和流行有其历史的原因。但是从技术角度来讲,这个文件格式是个非常差劲的格式。
GTF格式非常冗余。以人类转录组为例,Gencode V22的GTF文件为1.2G,压缩之后只有40M。大家知道压缩软件的压缩比是和软件的冗余程度。很少有文件能够压缩到1/30的大小。可见GTF格式多么冗余。GTF格式(及其早期版本GFF等)有很好的替代格式。从信息量上来讲:GTF 等价于 GenePred (Bed12) + Gene_Anno_table。
GenePred是Jimmy Kent创建UCSC genome browser的时候建立的文件格式。UCSC的文件格式定义是非常smart的,包括之后我可能会讲到的2bit,bigwig格式。
GTF vs GenePred
- 从文件大小上来看,压缩前:GTF(1.2G) >> Genepred(23M) + Gene_Anno_table (2.8M)。压缩后:GTF(40M) >> GenePred(7.8M) +Gene_Anno_table (580K)
- 从可读性来讲,GTF是以gene interval 为单位(行),每行可以是gene,transcript,exon,intron,utr等各种信息,看起来什么都在里面,很全面,其实可读性非常差,而且容易产生各种错误。GenePred格式是以transcript为单位,每行就是一个transcript,简洁直观。
- 从程序处理的角度来讲:以GTF文件作为输入的程序,如果换成以GenePred格式为输入,编程的难度会降低一个数量级,运算时间会快很多,代码的可读性强很多。
GTF 转换成GenePred:
gtfToGenePred -genePredExt -ignoreGroupsWithoutExons -geneNameAsName2 test.gtf test.gpd
Gene_Anno_table: 其实就是把GTF的所有transcript行的第9列转换变成一个表格。
MAF
MAF格式本来并不是一个常见的文本文件格式,只是因为癌症研究实在是太热门了,对它的理解也变得需求旺盛起来了。
MAF的说明
这些文件应该使用下面描述的突变注释格式(MAF)进行格式化。另外下文中有文件命名规范。
以下几种类型的体细胞突变会在MAF文件中出现: * 错义突变及无义突变 * 剪接位点,其定义为剪接位点2 bp以内的SNP * 沉默突变 * 与基因的编码区、剪接位点或遗传元件目标区域重叠的引物。 * 移码突变 * 调控区突变
大部分MAF提交提交的是原始数据。这些原始数据中在体细胞中标记的位点与已知的变异类型相重合的。为避免有可能出现的细胞系污染,MAF规定了一定的下细胞过滤标准。根据现行政策,可开放获取MAF资料应满足:
- 包括所有已验证的体细胞突变名称
- 包括与编码区域或剪接位点重叠的所有未验证的体细胞突变名称
- 排除所有其他类型的突变(即非体细胞突变、不在编码区域或剪接位点的未验证体的细胞突变以及未在dbSNP、COSMIC或OMIM中注释为体细胞的dbSNP位点)
我们提交给DCC MAF存档的数据包括两种:Somatic MAF(named .somatic.maf)的开放访问数据以及不经过筛选的包含原始数据的Protected MAF(named.protected.maf)。所有数据将使用MAF标准进行格式化。
MAF文件字段
MAF文件的格式是以制表符分隔的列。这些列都在表1中进行了详细注释,每个MAF文件中都必须按照相应格式进行处理,DCC将验证每列的顺序是否符合标注。每列的标题和值有时需要区分大小写。列中允许出现空值(即空白单元格)或枚举值。验证器将查找是否存在一个符合规范(例如#version 2.4)的标题,如果没有,验证将会失败。除了出现在表1中的列外,也可以选择附加其他列。可选列不经过DCC验证,可以按任何顺序进行。
MAF文件可能有两种格式 ,可能是47列,或者120列,第一行一般都是 头文件,注释着每一列的信息,的确,信息量有点略大。如下:
1 Hugo_Symbol
2 Entrez_Gene_Id
3 Center
4 NCBI_Build
5 Chromosome
6 Start_Position
7 End_Position
8 Strand
9 Consequence
10 Variant_Classification
11 Variant_Type
12 Reference_Allele
13 Tumor_Seq_Allele1
14 Tumor_Seq_Allele2
15 dbSNP_RS
16 dbSNP_Val_Status
17 Tumor_Sample_Barcode
18 Matched_Norm_Sample_Barcode
19 Match_Norm_Seq_Allele1
20 Match_Norm_Seq_Allele2
21 Tumor_Validation_Allele1
22 Tumor_Validation_Allele2
23 Match_Norm_Validation_Allele1
24 Match_Norm_Validation_Allele2
25 Verification_Status
26 Validation_Status
27 Mutation_Status
28 Sequencing_Phase
29 Sequence_Source
30 Validation_Method
31 Score
32 BAM_File
33 Sequencer
34 t_ref_count
35 t_alt_count
36 n_ref_count
37 n_alt_count
38 HGVSc
39 HGVSp
40 HGVSp_Short
41 Transcript_ID
42 RefSeq
43 Protein_position
44 Codons
45 Hotspot
46 cDNA_change
47 Amino_Acid_Change
1 Hugo_Symbol
2 Entrez_Gene_Id
3 Center
4 NCBI_Build
5 Chromosome
6 Start_Position
7 End_Position
8 Strand
9 Variant_Classification
10 Variant_Type
11 Reference_Allele
12 Tumor_Seq_Allele1
13 Tumor_Seq_Allele2
14 dbSNP_RS
15 dbSNP_Val_Status
16 Tumor_Sample_Barcode
17 Matched_Norm_Sample_Barcode
18 Match_Norm_Seq_Allele1
19 Match_Norm_Seq_Allele2
20 Tumor_Validation_Allele1
21 Tumor_Validation_Allele2
22 Match_Norm_Validation_Allele1
23 Match_Norm_Validation_Allele2
24 Verification_Status
25 Validation_Status
26 Mutation_Status
27 Sequencing_Phase
28 Sequence_Source
29 Validation_Method
30 Score
31 BAM_File
32 Sequencer
33 Tumor_Sample_UUID
34 Matched_Norm_Sample_UUID
35 HGVSc
36 HGVSp
37 HGVSp_Short
38 Transcript_ID
39 Exon_Number
40 t_depth
41 t_ref_count
42 t_alt_count
43 n_depth
44 n_ref_count
45 n_alt_count
46 all_effects
47 Allele
48 Gene
49 Feature
50 Feature_type
51 One_Consequence
52 Consequence
53 cDNA_position
54 CDS_position
55 Protein_position
56 Amino_acids
57 Codons
58 Existing_variation
59 ALLELE_NUM
60 DISTANCE
61 TRANSCRIPT_STRAND
62 SYMBOL
63 SYMBOL_SOURCE
64 HGNC_ID
65 BIOTYPE
66 CANONICAL
67 CCDS
68 ENSP
69 SWISSPROT
70 TREMBL
71 UNIPARC
72 RefSeq
73 SIFT
74 PolyPhen
75 EXON
76 INTRON
77 DOMAINS
78 GMAF
79 AFR_MAF
80 AMR_MAF
81 ASN_MAF
82 EAS_MAF
83 EUR_MAF
84 SAS_MAF
85 AA_MAF
86 EA_MAF
87 CLIN_SIG
88 SOMATIC
89 PUBMED
90 MOTIF_NAME
91 MOTIF_POS
92 HIGH_INF_POS
93 MOTIF_SCORE_CHANGE
94 IMPACT
95 PICK
96 VARIANT_CLASS
97 TSL
98 HGVS_OFFSET
99 PHENO
100 MINIMISED
101 ExAC_AF
102 ExAC_AF_Adj
103 ExAC_AF_AFR
104 ExAC_AF_AMR
105 ExAC_AF_EAS
106 ExAC_AF_FIN
107 ExAC_AF_NFE
108 ExAC_AF_OTH
109 ExAC_AF_SAS
110 GENE_PHENO
111 FILTER
112 CONTEXT
113 src_vcf_id
114 tumor_bam_uuid
115 normal_bam_uuid
116 case_id
117 GDC_FILTER
118 COSMIC
119 MC3_Overlap
120 GDC_Validation_Status
重要标准
列表中每列的顺序最好与索引列相同。
标有“Case Sensitive“的列所有标题都需要区分大小写。
标有”Null“的列表示允许具有空值。
标有“Enumerated column”的列表示具有指定的值,比如“Enumerated value” 是"No"表示该列没有指定的值;其他值表示允许列出的具体值; “Set”表示该列的值来自指定的一组已知值(例如HUGO基因符号)
MAF文件检查
DCC 档案验证器将检查MAF文件的完整性。如果MAF文件中的任何一项出现错误,验证将失败:
1. 列标题文本(包括大小写)和顺序必须与表1完全一致
2. 表1中列出的列标题下的值不是空值时必须具有相应的值
3. 表1中指定为“Case Sensitive”的值必须区分大小写。
4. 如果列标题在规范中列出为具有枚举值(即“Enumerated”列的“Yes”),则这些列中的值必须来自“Enumerated”下列出的枚举值。
5. 如果规范中列标题具有设置值(即“Enumerated”列的“Set”),则那些列下的值必须来自该域的枚举值(例如,HUGO基因符号)。
6. 所有Allele-based列必须包含 –(deletion)或由以下大写字母组成的字符串:A,T,G,C。
7. 如果Validation_Status == "Untested" 那么Tumor_Validation_Allele1, Tumor_Validation_Allele2, Match_Norm_Validation_Allele1, Match_Norm_Validation_Allele2 可以是空值(由 Validation_Status决定).
a) 如果Validation_Status == "Inconclusive" 那么Tumor_Validation_Allele1, Tumor_Validation_Allele2, Match_Norm_Validation_Allele1, Match_Norm_Validation_Allele2 可以是空值(由 Validation_Status决定)
8. 如果Validation_Status == Valid, 那么Validated_Tumor_Allele1 and Validated_Tumor_Allele2一定要是 A, C, G, T, 或“-“中的一种
a) 如果Validation_Status == "Valid" 那么Tumor_Validation_Allele1, Tumor_Validation_Allele2, Match_Norm_Validation_Allele1, Match_Norm_Validation_Allele2 不可以是空值
b) 如果Validation_Status == "Invalid" 那么Tumor_Validation_Allele1, Tumor_Validation_Allele2, Match_Norm_Validation_Allele1, Match_Norm_Validation_Allele2 不可以是空值Tumor_Validation_Allelle1 == Match_Norm_Validation_Allele1
Tumor_Validation_Allelle2 == Match_Norm_Validation_Allele2 (出现错误时,增加以替代8a)
9. 检查Mutation_Status的等位基因值:
检查Validation_status的等位基因值:
a) 如果Mutation_Status == "Germline" and Validation_Status == "Valid", 那么Tumor_Validation_Allele1 == Match_Norm_Validation_Allele1 Tumor_Validation_Allele2 == Match_Norm_Validation_Allele2.
b) 如果Mutation_Status == "Somatic" ,Validation_Status == "Valid", 那么Match_Norm_Validation_Allele1 == Match_Norm_Validation_Allele2 == Reference_Allele 且(Tumor_Validation_Allele1 or Tumor_Validation_Allele2) != Reference_Allele
c) 如果Mutation_Status == "LOH" and Validation_Status=="Valid", 那么Tumor_Validation_Allele1 == Tumor_Validation_Allele2 Match_Norm_Validation_Allele1 != Match_Norm_Validation_Allele2 and Tumor_Validation_Allele1 == (Match_Norm_Validation_Allele1 or Match_Norm_Validation_Allele2).
10. 检查 Start_position <= End_position
11. 根据Variant_Type检查Start_position和End_position:
a) 如果Variant_Type == "INS", 那么(End_position - Start_position + 1 == length (Reference_Allele) 或End_position - Start_position == 1) 且length(Reference_Allele) <= length(Tumor_Seq_Allele1 and Tumor_Seq_Allele2)
b) 如果Variant_Type == "DEL", 那么End_position - Start_position + 1 == length (Reference_Allele),
且length(Reference_Allele) >= length(Tumor_Seq_Allele1 and Tumor_Seq_Allele2)
c) 如果Variant_Type == "SNP", 那么length(Reference_Allele and Tumor_Seq_Allele1 and Tumor_Seq_Allele2) == 1 且(Reference_Allele and Tumor_Seq_Allele1 and Tumor_Seq_Allele2) != "-"
d) 如果Variant_Type == "DNP", 那么length(Reference_Allele 和Tumor_Seq_Allele1 and Tumor_Seq_Allele2) == 2 且(Reference_Allele and Tumor_Seq_Allele1 andTumor_Seq_Allele2) !contain "-"
e) 如果Variant_Type == "TNP", 那么length(Reference_Allele and Tumor_Seq_Allele1 and Tumor_Seq_Allele2) == 3 且(Reference_Allele and Tumor_Seq_Allele1 and Tumor_Seq_Allele2) !contain "-"
f) 如果Variant_Type == "ONP", 那么length(Reference_Allele) == length(Tumor_Seq_Allele1) == length(Tumor_Seq_Allele2) > 3 且(Reference_Allele and Tumor_Seq_Allele1 and Tumor_Seq_Allele2) !contain "-"
12. 基于UUID的文件的验证:
a) 列#33必须包含肿瘤样本的BCR等分试样的UUID的Tumor_Sample_UUID
b) 列#34必须是Matched_Norm_Sample_UUID,其中包含用于匹配正常样本的BCR等份的UUID
c) 由Tumor_Sample_Barcode和Matched_Norm_Sample_Barcode表示的元数据应分别对应于分配给Tumor_Sample_UUID和Matched_Norm_Sample_UUID的UUID
MAF命名协议
在上传到DCC的档案中,MAF文件名应与以下方式与包含档案名称相关: 如果存档名称是:
<domain>_<disease_abbrev>.<platform>.Level_2.<serial_index>.<revision>.0.tar.gz
那么具有存档的公开的MAF文件命名依据为:
<domain>_<disease_abbrev>.<platform>.Level_2.<serial_index>[.<optional_tag>].somatic.maf
具有存档的受保护的MAF文件命名依据为:
<domain>_<disease_abbrev>.<platform>.Level_2.<serial_index>[.<optional_tag>].protected.maf
<optional_tag>可能包含字母数字字符、破折号或下划线,但不能有空格或句号。<optional_tag>可以省略。使用<optional_tag>的目的是给出一些简短的注释。
例如,对于文件
genome.wustl.edu_OV.IlluminaGA_DNASeq.Level_2.7.6.0.tar.gz
有效的MAF名称的为:
genome.wustl.edu_OV.IlluminaGA_DNASeq.Level_2.7.preliminary.somatic.maf
genome.wustl.edu_OV.IlluminaGA_DNASeq.Level_2.7.protected.maf
参考链接:
https://wiki.nci.nih.gov/display/TCGA/Mutation+Annotation+Format+%28MAF%29+Specification+-+v2.4
https://software.broadinstitute.org/software/igv/MutationAnnotationFormat
https://wiki.nci.nih.gov/display/TCGA/Mutation+Annotation+Format+%28MAF%29+Specification